超高压液相色谱-串联质谱法测定奶粉中抗吸虫药物残留
2023-11-10杨帆张晓梅阎炳杨卫海孙静文李梓昱黄玲利张鸿伟徐杰
杨帆 张晓梅 阎炳 杨卫海 孙静文 李梓昱 黄玲利张鸿伟*, 徐杰*
1(中国海洋大学食品科学与工程学院,青岛266520)
2(青岛海关技术中心,青岛 266109)
3(青岛海关,青岛 266002)
4(华中农业大学动物科学技术学院、动物医学院,武汉 430070)
吸虫病是牛和羊等食草动物严重的寄生虫病之一,也是人畜共患的寄生虫病[1],给牲畜和人类健康带来巨大威胁,也会对畜牧业生产造成严重的经济损失[2]。在牛羊吸虫病的防治中,氯氰碘柳胺[3]、硝碘酚腈[4]、碘醚柳胺[5]和三氯苯达唑[6]为常用广谱驱虫药,对多种吸虫病有良好疗效[7]。抗吸虫药在我国牛羊养殖中普遍使用[8],但在停药期的不合理使用、动物产品生产加工不规范和环境污染等情况下[9],容易导致该类药物在牛羊乳中的残留[10]。目前研究发现,多数抗吸虫药具有较强的细胞毒性,而奶粉是牛羊乳的主要加工制品[11],长期食用含有抗吸虫药物残留的奶粉易导致人体产生抗药性,给消费者带来健康隐患[12]。当前,中国(GB 31650-2019)[13]和欧盟(EU(No)37/2010)[14]针对乳品中常见抗吸虫类药物均制定了最大残留限量(Maximum residue limits,MRLs),范围在10~45 μg/kg 之间,相关药物的化学结构和化学信息见图1。
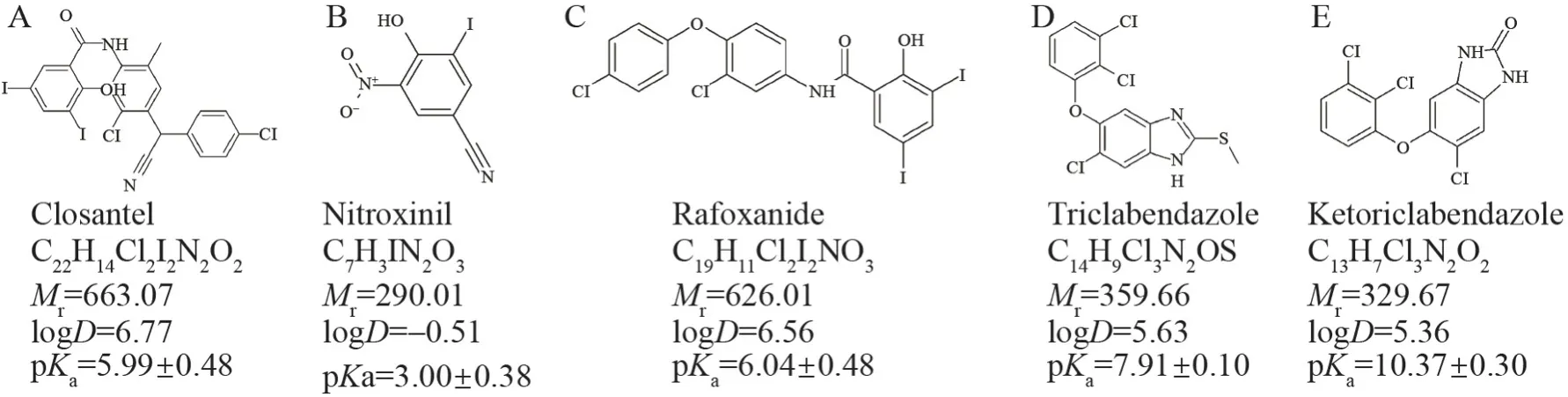
图1 氯氰碘柳胺(A)、硝碘酚腈(B)、碘醚柳胺(C)、三氯苯达唑(D)和三氯苯达唑酮(E)的化学结构和理化参数Fig.1 Chemical structures and physicochemical parameters of closantel (A),nitroxinil (B),rafoxanide (C),triclabendazole (D) and ketotriclabnedazole (E)
目前,我国尚未建立涵盖奶粉基质的抗吸虫药物残留检测方法标准,因此,建立灵敏可靠的分析方法具有现实紧迫性。相比普通液体乳,奶粉基质组成更为复杂[15],因此测定分析时,样品的前处理净化步骤显得尤为关键[16]。常用于抗吸虫药物残留测定的前处理净化方法有液-液萃取法[17]、固相萃取法[18]和QuEChERS 法[19]等,其中,固相萃取法和QuEChERS 法具有回收率高[20]、溶剂使用量少、操作简便、方法重现性好和易于标准化等优点,是目前应用较多的前处理净化方法[21]。常用于抗吸虫药物残留测定的分析方法有紫外-可见分光光度法[22]、胶体金免疫层析法[23]、薄层色谱法[24]、高效液相色谱法[25]、液相色谱荧光检测法[26]和液相色谱-串联质谱法(HPLC-MS/MS)[27]等。其中,HPLC-MS/MS 法具有选择性好[28]、灵敏度高[29]和定性与定量分析准确等优点[30],是目前抗吸虫药物残留的主要分析技术[31],但检测样品多集中于液态牛羊奶产品,如Devreese 等[32]基于固相萃取净化建立了HPLC-MS/MS 测定液态牛羊奶中氯氰碘柳胺残留的方法,检出限为10 μg/kg;李小桥[33]等基于液液萃取建立了超高效液相色谱-串联质谱(UPLC-MS/MS)测定液态牛奶基质中三氯苯达唑及其代谢物三氯苯达唑酮残留的方法,检出限为1 μg/kg。目前,针对奶粉基质的相关高通量检测方法研究仍鲜有报道。
本研究基于UHPLC-MS/MS,针对牛奶粉中常见抗吸虫药物(氯氰碘柳胺、硝碘酚腈、碘醚柳胺、三氯苯达唑及其代谢物三氯苯达唑酮)前处理净化的关键环节,考察了多种商业化净化材料(包括Captiva EMR-lipid 固相萃取柱、Captiva ND lipids 固相萃取柱、Cleanert LipoNo 除脂管和QuEChERS 净化管)的使用效果,并优化了色谱分离条件和质谱分析参数,建立了适用于检测奶粉中抗吸虫药物残留的高通量分析方法。
1 实验部分
1.1 仪器与试剂
APUS PLUS 超高压液相色谱仪(珂睿公司);Triple Quad 6500+三重四极杆质谱仪(美国SCIEX 公司);CR22N 型高速冷冻离心机(日本日立公司);Turbo Vap®LV 型高通量水浴氮吹浓缩仪(美国Caliper 公司)。
乙腈和甲醇(质谱纯,德国Merck 公司);甲酸(质谱纯,美国Fluka 公司);甲酸铵(质谱纯,德国Sigma 公司);氨水、NaCl 和无水MgSO4(分析纯,国药集团化学试剂有限公司)。Captiva EMR-lipid 固相萃取柱(3 mL/300 mg,美国Agilent 公司)和Captiva ND lipids 固相萃取柱(3 mL/60 mg,美国Agilent 公司);Cleanert LipoNo 除脂管(15 mL,天津博纳艾杰尔公司);N-丙基乙二胺(Primary secondary amine,PSA)和十八烷基硅烷键合硅胶(C18,美国Agilent 公司)。实验用水为超纯水(电阻率≥18.2 MΩ·cm)。
氯氰碘柳胺(CAS 登录号:57808-65-8,纯度98.1%)和硝碘酚腈(CAS 登录号:1689-89-0,纯度98.8%)购于德国Dr.Ehrenstorfer 公司;碘醚柳胺(CAS 登录号:22662-39-1,纯度96.0%)和三氯苯达唑酮(CAS 登录号:1201920-88-8,纯度99.4%)购于加拿大Toronto Research Chemicals 公司;三氯苯达唑(CAS 登录号:68786-66-3,纯度98.8%)购于英国Laboratory of the Government Chemist 公司。分别称取10 mg 标准品,用乙腈溶解并定容至10 mL,制备1 g/L 标准储备溶液,于–20 ℃保存备用(此条件下可保存6 个月)。分别移取100 μL 相应标准储备液于10 mL 容量瓶中,用乙腈定容,制备成混合标准中间溶液(10 mg/L),于–20 ℃保存备用(此条件下可保存1 个月)。混合标准工作液(1 mg/L)用乙腈-水(7∶3,V/V)溶液配制,现用现配。
1.2 样品前处理
称取(2.00±0.02)g 牛奶粉于离心管中,用4 mL 水溶解,混匀,加入16 mL 冰乙腈(将乙腈于–20 ℃预冷3 h),涡旋振荡10 min,以10000 r/min 离心10 min,取上清液备用。
Captiva ND 法净化流程 取10 mL 备用上清液于离心管中,加2 g NaCl,涡旋振荡10 min,以10000 r/min 离心10 min,取5 mL 上清液,于40 ℃下氮吹至近3 mL,过Captiva ND lipids 固相萃取柱并抽干,收集滤出液,氮吹至近干。
Captiva EMR 法净化流程 取5 mL 备用上清液,过Captiva EMR lipid 固相萃取柱并抽干,再用2 mL乙腈洗脱,收集全部滤出液,于40 ℃下氮吹至近干。
Cleanert LipoNo 法净化流程 向上清液中加入2 g 无水MgSO4和0.5 g NaCl,涡旋振荡5 min,以5000 r/min 离心3 min,取5 mL 上清液于Cleanert LipoNo 除脂萃取管中,涡旋振荡5 min,以5000 r/min离心3 min,取2.5 mL 上清溶液,于40 ℃下氮吹至近干。
QuEChERS 法净化流程 向离心管中加入4 g MgSO4和1 g NaCl,涡旋振荡3 min,以10000 r/min 离心5 min,取8 mL 上清液转移至装有净化剂的离心管(含50 mg PSA 和150 mg C18)中,涡旋振荡5 min,以10000 r/min 离心5 min,取5 mL 上清溶液,于40 ℃下氮吹至近干。
将以上4 种氮吹至近干的提取残渣用500 μL 乙腈-水(7:3,V/V)溶液复溶,涡旋并超声1 min,15000 r/min 离心10 min,取上清液进行UHPLC-MS/MS 测定。
1.3 仪器条件
色谱条件 Waters CORTECS UPLC C18色谱柱(50 mm×3.0 mm,1.6 μm)。流动相A 为5 mmol/L 甲酸铵溶液,流动相B 为乙腈。梯度洗脱程序:0~0.1 min,15%B;0.1~2.2 min,15%~100%B;2.2~4.0 min,100%B;4.0~4.2 min,100%~15%B;4.2~5.5 min,15%B。自动进样器温度:15 ℃;流速:0.4 mL/min;进样体积:5 μL;柱温:30 ℃。
质谱条件 电喷雾电离(ESI)采用负离子模式;离子化电压:–4500 V;气化温度:575 ℃;气帘气压力:45.0 kPa(N2);喷雾气压力:45 kPa(N2);辅助加热气压力:65 kPa(N2);碰撞气:Medium(N2);检测模式:多反应监测(MRM)模式。
目标分析物的色谱-质谱参数见表1。

表1 抗吸虫药物的化学信息及相关分析参数Table 1 Chemical information and analytical parameters of anthelmintics
1.4 基质效应评价
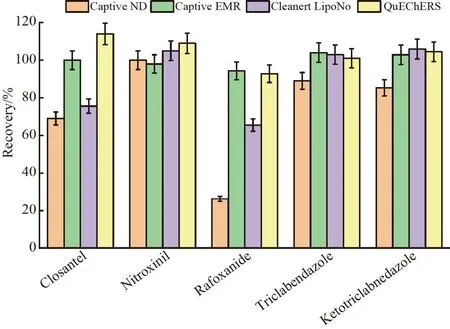
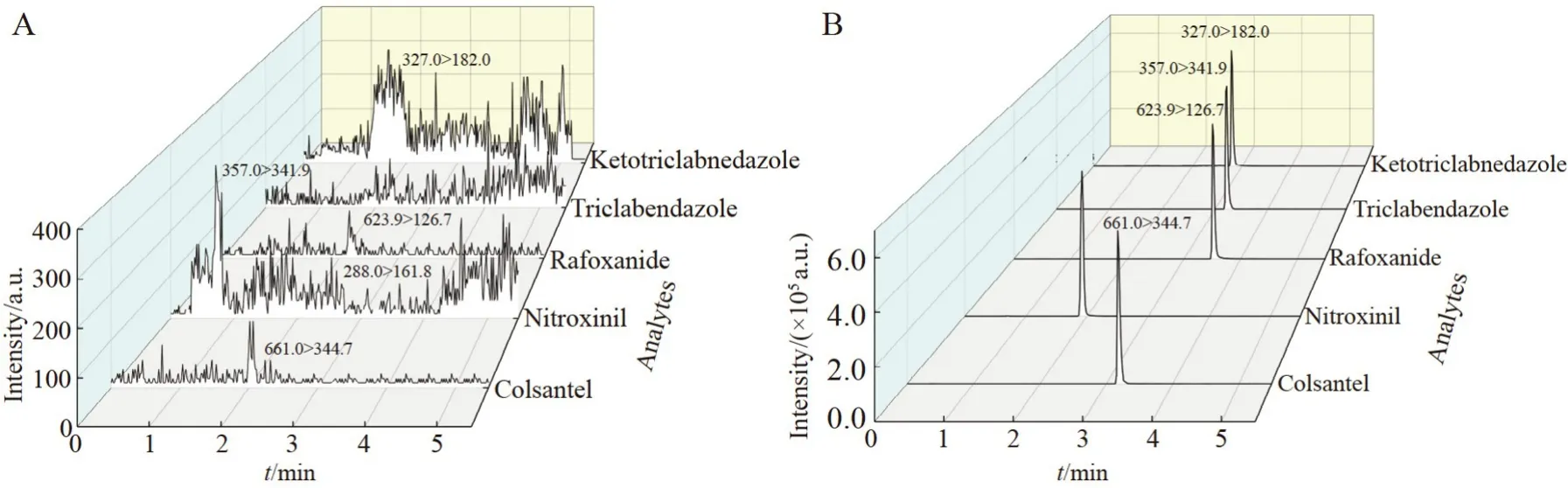
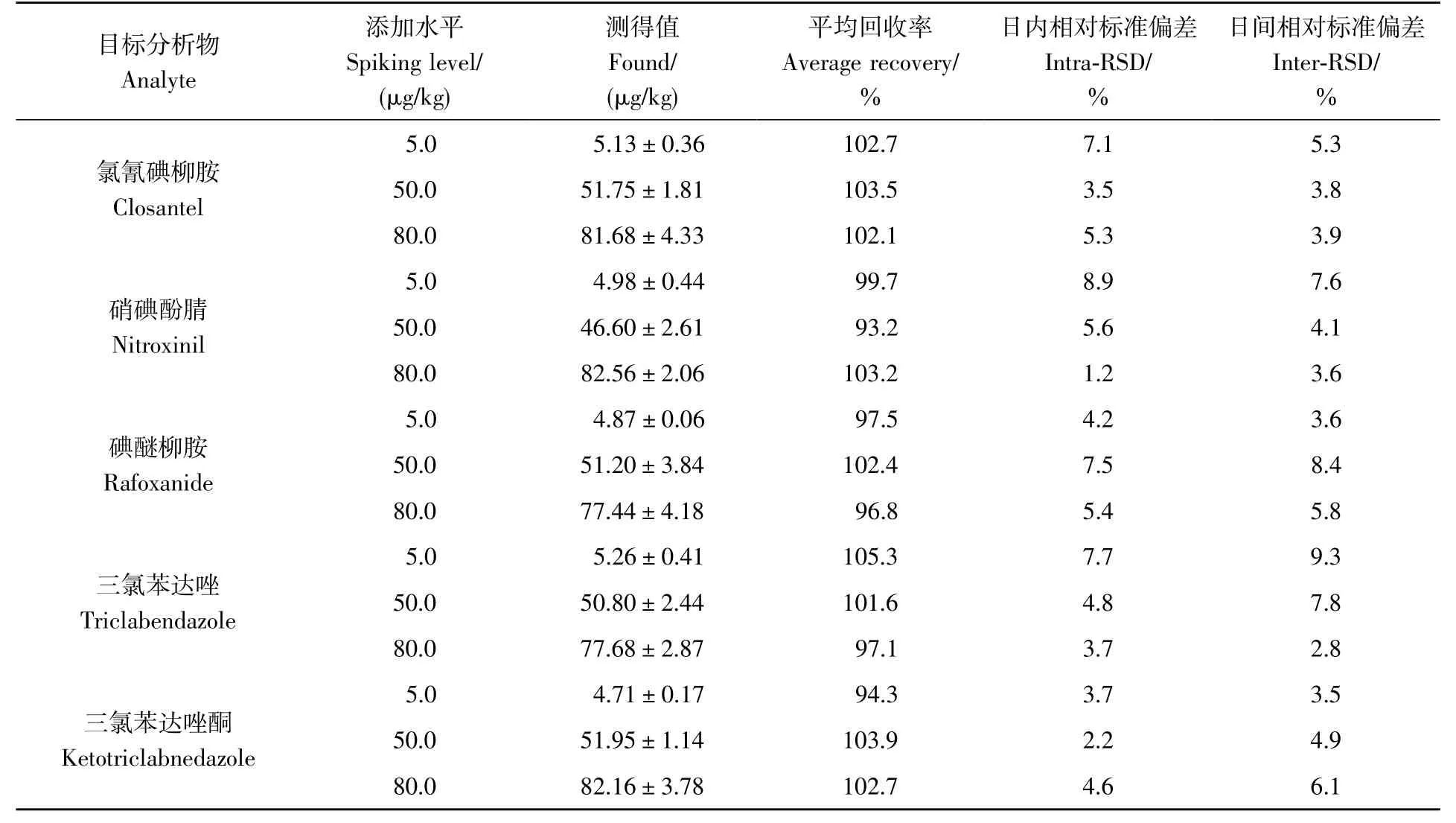
基质效应(Matrix effect,ME)的计算方法:ME=基质标准曲线斜率/溶剂标准曲线斜率。若ME <0.8,说明存在基质抑制效应;若ME >1.2,说明存在基质增强效应;若0.8 奶粉基质成分复杂,含有大量蛋白质、脂质和碳水化合物等大分子,还可能含有低聚糖、不饱和脂肪酸和微量元素等营养成分,在前处理过程中需将这些干扰物有效去除。根据目标分析物的极性特点,选用提取后不通过乳化作用即可沉降蛋白质的乙腈作为提取溶剂。在低温条件下,蛋白质结构相对稳定,不易与目标分析物结合而形成共沉淀,因此,采用预冷的冰乙腈可以有效去除蛋白质。 为更好地提升方法的分析性能和检测通量,本研究针对牛奶粉基质的特点和目标分析物的理化性质,考察了4 种快速净化固相萃取材料(Captiva ND lipids 固相萃取柱、Captiva EMR lipids 固相萃取柱、Cleanert LipoNo 除脂管和QuEChERS 净化管)对空白奶粉基质添加目标分析物(10 μg/kg)的回收率的影响,结果见图2。 图2 不同净化方法对空白基质添加(10 μg/kg)5 种目标分析物回收率的影响(n=3)Fig.2 Effects of different clean-up methods on spiking recoveries of 5 kinds of analytes at spiking concentration level of 10 μg/kg (n=3) 采用Cleanert LipoNo 除脂萃取管和Captiva ND lipids 固相萃取柱时,Closantel 和Rafoxanide 的回收率较低,推测其主要是因为Closantel 和Rafoxanide 的分配系数较大,具有较强的非极性,在相应填料上形成了非选择性吸附而不易被洗脱。相比之下,基于QuEChERS 法和Captiva EMR lipids 固相萃取柱净化的方法针对5 种目标分析物都具有比较好的回收率,并且在空白奶粉基质中添加(10 μg/kg)回收率的差异性不显著(p>0.05)。其中,QuEChERS 法采用的填料为PSA 和C18,PSA 具有较强的离子交换能力,常用于吸附糖和色素等添加剂,C18常用于吸附脂肪等含长碳链的中等极性到非极性的化合物,因此吸附奶粉基质组分的能力较强,而碳链较短的目标分析物可以更好地被洗脱,因此整体回收率较高。Captiva EMR lipids 固相萃取柱是基于体积排阻和疏水相互作用机制去除脂质,在有效去除奶粉基质中磷脂的同时,可最大程度地减少离子抑制对目标分析物的影响,整体回收率也较好。对比QuEChERS 法,EMR 法采用滤过式净化方式可有效缩短前处理时长,并且产生的废物较少。综合考虑回收率、处理通量和环境友好等因素,本研究最终选用Captiva EMR lipids 固相萃取柱作为净化材料。此外,根据目标分析物的极性,优化了定容溶剂的组成,最终选用乙腈-水(7∶3,V/V)作为定容溶剂。 由于目标分析物同时含有氨基和氯离子,因此,目标物在电喷雾电离负离子模式(ESI-)和正离子模式(ESI+)下均可以进行离子化,结果表明,ESI-模式下各目标分析物的电离效果较好,同时考虑到ESI-的背景响应低,因此选择ESI-条件进行测定。采用混合标准工作液优选各目标化合物的碎片离子、优化碰撞能量和去簇电压等质谱参数(表1)。本研究选用CORTECS UPLC C18色谱柱(50 mm×3.0 mm,1.6 μm),该柱分离效能好,在适中pH 值范围内可实现稳定的酸、碱和中性化合物保留,短柱长有利于提升检测通量。本研究选用乙腈-水流动相体系,并在水相中加入5 mmol/L 甲酸铵改善峰形,以减小拖尾和双峰现象,通过优化梯度洗脱条件,可在5.5 min 内完成目标分析物的1 个测定循环。 在优化的色谱-质谱参数(表1)条件下,5 种目标分析物在空白基质溶液中和基质匹配定量限水平(2.5 μg/kg)标准溶液中的提取离子流图见图3A 和3B,在目标分析物的保留时间窗口内,基线丰度得到了有效控制。 图3 (A)空白基质溶液和(B)2.5 μg/kg 基质匹配标准溶液中5 种目标分析物的提取离子流图Fig.3 Extracted ion chromatograms of 5 kinds of analytes in(A)matrix blank solution and(B)matrix-matched standard solution (2.5 μg/kg) 2.3.1 方法的基质效应、线性范围、检出限和定量限 使用已经过确证不含5 种目标分析物的市售奶粉样品作为本研究的空白基质。采用此空白基质绘制基质匹配标准曲线,配制浓度水平分别为1.0、5.0、10.0、20.0、50.0 和100.0 μg/L 的基质匹配标准溶液,以目标分析物浓度为横坐标、定量离子提取离子流色谱峰面积为纵坐标,最小二乘法回归分析拟合标准曲线,得到各目标分析物的回归方程和相关系数(r)。以标准曲线法计算3 倍信噪比(S/N=3)时的添加浓度为方法检出限(Limit of detection,LOD),10 倍信噪比(S/N=10)时的添加浓度为定量限(Limit of quantification,LOQ),结果表明,各目标分析物的LOQ 远小于其各自的MRLs(表2)。 表2 5种目标分析物的线性方程、相关系数、检出限、定量限和基质效应Table 2 Linear equations,coefficients of correlation (r),limits of detection (LODs),limits of quantification (LOQs) and matrix effects (MEs) of 5 kinds of analytes 对基质效应(MEs)的评估结果见表2。由表2 可见,本方法对各目标分析物存在一定程度的基质增强效应(1.2 2.3.2 回收率与精密度 本研究采用基质空白添加进行回收率与精密度实验。选择空白牛奶粉样品,依据5 种目标分析物的MRLs,选择3 个添加水平(5.0、50.0 和80.0 μg/kg),按照1.3 节所述的前处理步骤,相当于目标分析物的浓度水平为5.0、50.0 和80.0 μg/L。每个水平于同日做6 个平行,计算实际测定值、回收率和日内相对标准偏差(Intra-relative standard deviations,Intra-RSDs),每隔2 天重复1 次前述实验,重复3 次,计算日间相对标准偏差(Inter-RSDs)。本研究的平均回收率和RSD 结果见表3。 表3 奶粉样品中5种目标分析物的加标回收率和相对标准偏差Table 3 Recoveries and relative standard deviations (RSDs) of 5 kinds of analytes spiked in milk powder samples (n=6) 由表3 可知,目标分析物的平均回收率为93.2%~105.3%,日内相对标准偏差为1.2%~8.9%,日间相对标准偏差为2.8%~9.3%。在提取效率确定的情况下,影响方法回收率和精密度等关键性能指标的主要因素为样品的前处理过程,本研究结果表明,本方法在保证高检测通量(快速净化)的同时也能保证性能指标的稳定。 采用本方法对30 份市售牛奶粉产品(10 个品牌的1、2、3 段奶粉)进行以上5 种目标化合物的检测,未在样品中检出抗吸虫药物残留。 为验证本方法的稳健性,对30 份不同日期不同分析批次的内部质控样品(在定量限水平进行空白基质添加的样品)进行平行测定(n=3)分析,回收率统计结果见电子版文后支持信息图S1。结果表明,5 种目标分析物的质控结果均未超出各自的1 倍标准偏差范围,说明本方法的稳健性良好。 本研究针对牛奶粉中常用抗吸虫药物残留,基于通过式固相萃取净化的方式建立了高通量UHPLCMS/MS 测定分析方法。本方法的提取步骤简单、快速,净化方法可以有效控制牛奶粉的基质效应,仪器分析时长控制在5.5 min 内,大幅提高了样品的分析通量。5 种常用抗吸虫药及其代谢物的方法定量限为2.61~4.95 μg/kg,日间精密度为2.8%~9.3%,指标满足相关药物的检测监测分析要求,可用于奶粉中相关药物残留的监控与监测以及日常检测。 支持信息 Supporting Information 图S1 30 批次质控奶粉样品中5 种目标分析物的定量测定质控图:(A)氯氰碘柳胺;(B)硝碘酚腈;(C)碘醚柳胺;(D)三氯苯达唑;(E)三氯苯达唑酮Fig S1 Quality control charts of 5 kinds of analyts in quality control samples within 30 different analysis batches: (A) Closantel;(B) Nitroxinil;(C) Rafoxanide;(D) Triclabendazole;(E) Ketotriclabnedazole2 结果与讨论
2.1 样品前处理条件的优化
2.2 仪器分析条件的优化
2.3 方法验证

2.4 方法的应用及稳健性评估
3 结论


