附聚/超支化阴离子色谱固定相的制备和性能研究
2023-11-10韩宇宁弘宇戴徐鑫黄忠平刘会君朱岩
韩宇 宁弘宇 戴徐鑫 黄忠平* 刘会君 朱岩
1(浙江工业大学化学工程学院,杭州 310014)
2(浙江大学化学系,杭州 310028)
1975 年,Small 等[1]首次提出抑制型离子色谱法,成功地解决了用电导检测器连续检测柱流出物的难题。抑制型离子色谱法被广泛应用于食品安全[2-5]、环境监测[6-8]和生物医药[9]等行业。抑制型离子色谱法具有快速、灵敏、可同时测定多组分等优点,可以测定很多目前难以用其它方法测定的离子尤其是阴离子。因此,开发新型阴离子色谱固定相以满足日益复杂的分离需求,已成为离子色谱领域的研究热点。离子色谱中影响选择性的关键因素是固定相,包括聚合物的基质组成[10]、离子交换基团的类型和结构等[11-12]。采用新的材料基质、新的修饰技术及更适宜的装填方式等方法提高色谱柱的柱效、选择性和分析速度等,已经成为该领域目前的主要研究方向。
聚电解质附聚型阴离子交换固定相是由阳离子聚电解质主链的功能基团通过静电作用吸附于带负电荷的基质表面制备得到[13-15]。阳离子聚电解质是一种水溶性高分子化合物,其链段带有大量可电离基团,因此兼具小分子电荷基团和高分子长链的双重结构特征[16]。尤其是季铵盐类高分子化合物,已经成为近年来的研究热点,发展较快、种类较多,是一类重要的功能性高分子聚电解质。Zhao 等[17]通过水热碳化法将蔗糖转化的碳质球作为基质,有效避免了有机聚合物基质与可极化阴离子之间的非离子相互作用,采用甲胺(MA)和1,4-丁二醇二缩水甘油醚(BDDE)制备聚电解质并接枝到表面。Zhang 等[18]通过巯基介导的聚合方法在聚苯乙烯-二乙烯基苯(PS-DVB)基质上引入氨基,采用2-(甲氨基)乙醇和BDDE 合成阳离子聚电解质,并与基质上的氨基反应,探究了巯基介导的聚合过程中形成的羧基/羟基中间层对固定相交换容量和疏水性的影响。超支化改性方法是在基质表面引入反应部位,然后通过缩合反应等方法接枝高支化聚合物,超支化层附着在基质表面,不穿透基底颗粒的孔隙,从而可以提高分离效率[19-22]。Uzhel 等[23]制备了3 种基于PS-DVB 的超支化阴离子交换剂,第一次循环分别采用甘氨酸、牛磺酸和乙醇胺与BDDE 反应,后4 次循环用甲胺(MA)与BDDE 反应,并比较了氨基取代基对固定相色谱性能的影响。Popov 等[24]采用PS-DVB 作为基质,在功能层结构中引入两性离子片段制备了共价键超支化阴离子色谱固定相,在25 min 内实现了7 种常见阴离子的分离。
附聚型离子色谱固定相结合超支化修饰方法是现代商用固定相最主要的修饰形式[25]。Pohl 等[26]采用二环氧试剂和伯胺(摩尔比1∶1)在柱内磺化基质上原位生成线性结构的聚电解质,再进行二环氧试剂和伯胺试剂交替循环反应形成超支化层,已应用于商业化色谱柱(如IonPac AS19-24)生产。近年来,研究者多尝试采用不同材质的固定相基质,或以不同的环氧和伯胺试剂制备聚电解质和超支化层,但未见类似柱内原位合成的研究报道。Huang 等[27]在PS-DVB 基质中嵌入氧化后的纳米金刚石,提高了基质微球的机械强度,后续以BDDE 和MA 制备阳离子聚电解质和超支化层制备阴离子柱,在20 min 内实现了7 种阴离子的基线分离。Huang 等[28]以环氧氯丙烷和MA 为单体制备季铵盐阳离子聚电解质和超支化层,15 min 内实现了5 种常规阴离子和3 种极化阴离子的分离。上述研究考察了固定相的选择性受基质类型和二环氧试剂的结构、超支化修饰次数等的影响,但阳离子聚电解质的合成条件及修饰量对柱效和选择性的影响尚未见报道。
柱容量、选择性和柱效是评价离子色谱固定相的3 个主要指标。离子色谱固定相的柱容量和选择性受洗脱条件、色谱柱温[29]、超支化层数以及基质表面官能团的化学性质[30-31]等因素的影响;柱效主要由固定相基质类型[32]、基质修饰方式[12]和装填方式[33]决定。本研究以PS-DVB 微球为基质,通过BDDE 和MA 制备阳离子聚电解质层,考察了聚电解质的合成条件及修饰量、基质微球的孔径和超支化次数对固定相分离度及柱效的影响,确定了最佳的合成条件。此外,对固定相的批间重现性及稳定性进行了考察。
1 实验部分
1.1 仪器与试剂
ICS-1100 离子色谱仪(美国Thermo Fisher Scientific 公司);ZD-10 装柱机(大连日普利公司);TG-16-WS 台式高速离心机(湖南湘仪公司);SK2510HP 超声波清洗仪(上海KUDOS 公司);SHZ-D Ⅲ循环水式真空泵(巩义予华公司);不锈钢柱(150 mm×4.6 mm,大连日普利公司)。
10 μm PS-DVB 微球(孔径分别为10 和30 nm,苏州纳微科技有限公司);BDDE(60%)、MA(40%)和冰醋酸(99.5%)购自上海阿拉丁公司;NaF、NaBr、NaNO2和NaNO3(99%)购自上海麦克林公司;Na3PO4(99%)购自宜兴第二化学试剂厂;(NH4)2SO3(99%)购自上海五四化学试剂厂;Na2CO3(99.8%)购自太仓美达试剂公司;NaHCO3(99.5%)购自温州市化学用料厂。所用试剂均为分析纯;实验用水为去离子水。
1.2 实验方法
将购买的PS-DVB 微球乳液用循环水式真空泵抽滤,再用无水乙醇洗涤2~3 次,置于60 ℃烘箱中烘干,备用。
1.2.1 固定相的制备
参考文献[26]的方法合成阳离子聚电解质。配制40 mL BDDE(10%,m/V)溶液,加入250 mL 三口烧瓶中,水浴升温至60 ℃,用恒压漏斗在1 h 内滴加40 mL 甲胺(4%,m/V)溶液,滴加完成后冷却至室温。将制备的阳离子聚电解质以5000 r/min 离心5 min,弃去沉淀,备用。
参考文献[26]的方法对基质磺化和阳离子聚电解质的附聚接枝。取2.5 g PS-DVB 微球置于250 mL三口烧瓶中,加入15 mL 冰醋酸搅拌10 min。在30 ℃下加入3 mL 二氯甲烷溶胀,搅拌30 min。缓慢滴加7 mL 浓H2SO4,搅拌3 min 后将反应液倒入1 mol/L 冰的稀H2SO4中猝灭。将磺化微球用去离子水反复清洗至过滤液为中性。将磺化后的基质超声分散于100 mL 去离子水中,在40 ℃水浴中,加入80 mL阳离子聚电解质,恒温搅拌附聚24 h,用去离子水洗涤至过滤液呈中性。将微球超声分散于100 mL 去离子水中,置于60 ℃水浴中,倒入20 mL BDDE 水溶液反应30 min,用去离子水洗涤3 次。将微球重新分散于100 mL 去离子水中,置于60 ℃水浴中,加入20 mL 甲胺溶液,反应30 min,完成一次接枝反应。反应路线如图1 所示。
1.2.2 固定相的装填
将固定相分散于60 mL 去离子水中形成匀浆液,将匀浆液倒满匀浆罐,在20 MPa 压力下进行填充,匀浆液以及顶替液均为去离子水,待流出液为500 mL 时装柱停止,在30 min 内缓慢降低流速将压力降为0 MPa。将色谱柱拆下后,以2 mmol/L Na2CO3和2 mmol/L NaHCO3溶液为流动相,流速为1.0 mL/min,冲洗平衡6 h 后,备用。
1.2.3 标准溶液的配制
采用赛默飞世尔科技有限公司IonPac AS19-24 商品柱性能报告中的7 种典型阴离子(F–、Cl–、NO2–、Br–、NO3–、PO43–和SO42–)评价自制色谱柱的分离性能。称取相应质量的NaF、NaCl、NaNO2、NaBr、NaNO3、Na3PO4•10H2O 和(NH4)2SO4,配制成浓度为1000 mg/L 的储备液,于4 ℃保存备用。将各离子的单标溶液配制成相应浓度(F–为2 mg/L,Br–为20 mg/L,NO2–为10 mg/L,Cl–为5 mg/L,NO3–为20 mg/L,PO43–为30 mg/L,SO42–为10 mg/L)的混合标准溶液。
1.2.4 色谱条件及柱容量的测定
色谱条件 在未特别注明的情况下,采用的色谱柱均为不锈钢柱(150 mm×4.6 mm);流动相:2.0 mmol/L Na2CO3和2.0 mmol/L NaHCO3的混合溶液;流速:1.0 mL/min;进样量:25 μL;抑制电流:25 mA;柱温:30 ℃;检测器:电导检测器。
柱容量的测定 用1.0 mol/L NaCl 溶液在高压泵上以1.0 mL/min 流速冲洗色谱柱6 h,使所有季铵基吸附位点带上Cl–。采用去离子水以相同的流速冲洗色谱柱6 h,冲出柱中游离的Cl–。最后,采用2.0 mmol/L Na2CO3和2.0 mmol/L NaHCO3的混合液以1.0 mL/min 流速冲洗色谱柱6 h,并收集洗出液,用离子色谱检测洗出液中Cl–的含量。
2 结果与讨论
2.1 阳离子聚电解质的合成优化
在制备过程中,MA 与BDDE 反应时易发生凝胶结块现象,所得的阳离子聚电解质在与磺化基质附聚后结构不稳定,并且接枝两次后不能实现7 种常见阴离子的基线分离。通过调节恒压漏斗活塞控制MA 的滴加速率制备阳离子聚电解质,分别调整滴加速率使得甲胺溶液在0.5 和1.0 h 内加至BDDE 中。两种不同的MA 流速下制备的阳离子聚电解质均未发生凝胶结块的情况。以两种方式制备的阳离子聚电解的色谱分离图如图2 所示,经过附聚接枝后,两种色谱性能差异明显。基质微球附聚0.5 h 内滴加MA 制备的阳离子聚电解所得的固定相,可对7 种常规阴离子进行初步的分离,而在1.0 h 内滴加MA 制备的阳离子聚电解质所得的固定相对阴离子保留较弱。然而,通过后续的接枝修饰后,情况却发生了相反的变化,如图2A 所示,附聚后对阴离子有初步分离能力的固定相未能实现7 种阴离子的分离;但是,附聚后对阴离子保留较弱的固定相却在两次接枝后具有较好的选择性,实现了7 种阴离子的基线分离,如图2B 所示。在1.0 h 内滴加MA 控制反应速率,使聚合的电解质呈线性生长,避免反应速率过快导致电解质聚合封闭成环。因此,后续实验采用在1.0 h 内滴加MA 制备阳离子聚电解质。

图2 7 种常规阴离子在固定相上分离的色谱图:(A)在0.5 h 内滴加40 mL MA 制备阳离子聚电解质制备的固定相;(B)在1 h 内滴加40 mL MA 制备阳离子聚电解质制备的固定相Fig.2 Chromatograms of separation of seven conventional anions on cationic polyelectrolyte agglomeration stationary phases prepared by dropping 40 mL of methylamine within(A) 0.5 h and (B) 1 h,respectively
2.2 不同附聚修饰量的固定相的色谱性能对比
采用不同量的阳离子聚电解质与磺化基质进行附聚及接枝修饰,并测试了所制备的固定相的色谱性能。如图3 所示,经过2 次接枝修饰反应后,采用不同量阳离子聚电解质附聚制备的固定相色谱性能相近,柱效都维持在同一水平。但是,Br–峰型不同,通过计算Br–峰的不对称因子(峰不对称因子:峰两侧峰高10%处距峰值位置的距离之比)可知,在采用40 mL 阳离子聚电解质附聚时,所制备的固定相对Br–的峰不对称因子为1.35,不对称因子较大。但是,当附聚的阳离子聚电解质体积达到80 mL 时,Br–峰型有所改善,不对称因子为1.12。磺化基质表面需要附聚足够量聚电解质,以消除基质对离子的作用力。聚电解质量为40 mL 时,附聚量不足,使得部分微球基质表面暴露于流动相中,导致Br–峰的不对称因子较大。当聚电解质量增大至80 和120 mL 时,色谱柱性能没有变化,说明磺化基质表面已经有足够量的聚电解质进行附聚。因此,后续研究采用80 mL 阳离子聚电解质附聚在磺化的基质上。

图3 采用(A)40 mL、(B)80 mL 和(C)120 mL 阳离子聚电解质附聚制备的固定相的色谱分离图Fig.3 Chromatograms of stationary phase prepared by attachment of 40 mL(A),80 mL (B) and 120 mL(C) of cationic polyelectrolyte
2.3 不同孔径基质的色谱性能
考察了10 nm 孔径的10 μm 微球与30 nm 孔径的10 μm 微球基质在相同条件下制备的固定相的色谱性能,结果如图4 所示,两种基质的固定相均能实现7 种常规阴离子的基线分离。在相同的流动相条件下,采用孔径10 nm 的基质微球制备的固定相对常规阴离子的分离度更大,但是保留时间过长;而孔径30 nm的基质微球可在20 min 内实现分离,相较孔径10 nm 的基质微球具有更快的分析速度。原因在于孔径10 nm 的基质微球比孔径30 nm 的基质微球具有更大的比表面积,能够附聚更多量的阳离子聚电解质,因此同样量的固定相其交换容量越大,保留能力越强。后续研究均采用孔径30 nm 微球基质制备固定相。

图4 (A)孔径30 nm 和(B)孔径10 nm 微球制备的基质色谱性能的对比Fig.4 Comparison of chromatographic performance of different stationary phase matrices: (A) Pore size of 30 nm;(B) Pore size of 10 nm
2.4 不同接枝次数的固定相色谱性能对比
阳离子聚电解质通过静电力与范德华力作用附聚于磺化的PS-DVB 微球基质的表面,形成类似核壳结构的薄膜[13-14],而离子交换固定相的功能基团是季铵基,微球表面的季铵功能基团的数量可以通过接枝次数控制,因此本研究考察了不同接枝修饰次数固定相的色谱性能。如图5 所示,接枝2 次和3 次的固定相具有良好的选择性,接枝3 次的固定相柱容量为0.43 mmol/column,高于接枝2 次的固定相(0.27 mmol/column)。当接枝4 次时,固定相柱容量上升至0.54 mmol/column,但选择性下降,NO3–与PO43–无法达到基线分离。接枝5 次后固定相的选择性进一步下降。推测是随着接枝次数增加,基质表面季铵功能基团数量增多,基质表面的电荷密度变小,造成柱效与选择性下降[34-35]。后续研究均对附聚后的微球基质接枝2 次制备固定相。

图5 不同接枝次数的固定相色谱性能的对比Fig.5 Comparison of chromatography performances of stationary phases with different numbers of grafting
2.5 固定相的评价
2.5.1 固定相的批次间重现性
在确定了色谱固定相合成的最佳条件后,考察了批次间合成的重现性。在2 个月内合成了8 批次的柱填料,并分别按照相同步骤进行色谱柱的装填,采用7 种常规阴离子的混合标准溶液进行色谱性能测试,结果如图6 与表1 所示,所有批次的柱填料均在相同色谱条件下测试,固定相的背景压力均小于4.83 MPa。对于SO42–,所有批次固定相的柱效均大于30000 N/m,峰不对称因子在0.98~1.24 之间,保留时间在22.71~25.85 min 之间。因此,本研究中的固定相的合成与填装方法的重现性良好,具有实现产业化的基础。

表1 不同批次固定相的色谱性能数据Table 1 Chromatographic performance data of different batches of stationary phase
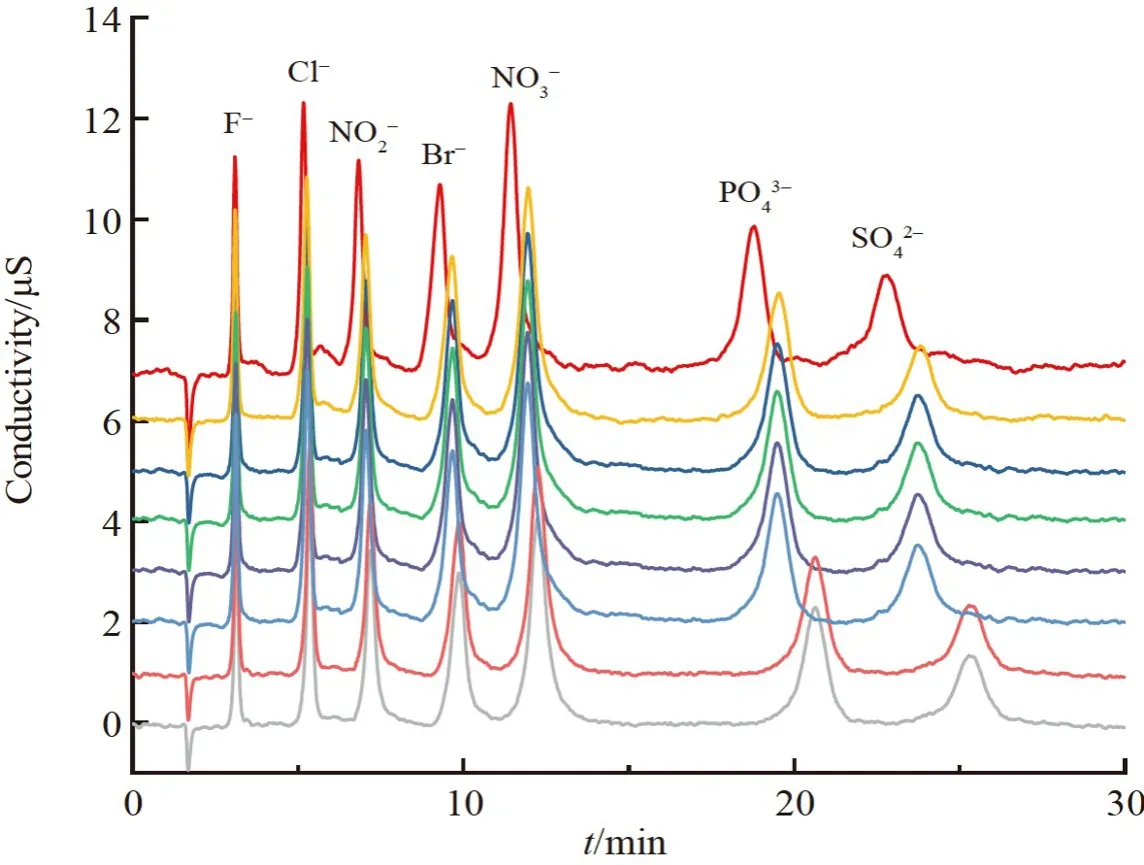
图6 不同批次固定相的色谱分离图Fig.6 Chromatographic separation diagrams of different batches of stationary phases
2.5.2 固定相的稳定性
在相同的色谱条件下,采用7 种常规阴离子混合标准液进行了100 次分离测试,并每隔4 针选取1 针分析结果进行色谱图的叠加绘制,如图7 所示,各离子保留时间的RSD 均不超过1.1%,峰高与峰面积的RSD 均小于6%,表明此固定相具有良好的稳定性和耐久性。
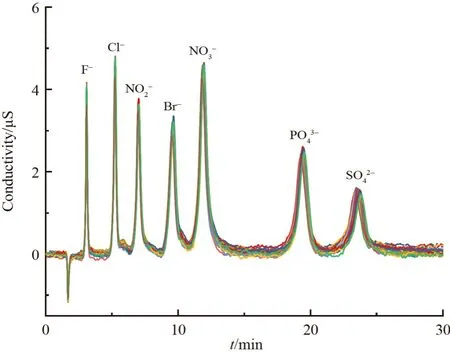
图7 7 种常见阴离子在固定相上的分离叠加图Fig.7 Separation and superimposition diagram of 7 kinds of common anions on the stationary phase
3 结论
考察了影响附聚型阴离子色谱固定相柱效和分离度的因素,解决了聚阳离子电解质在合成过程中容易产生凝结及难以重现的问题,探究了聚阳离子电解质对固定相柱效及分离度的影响。优化后的固定相性能良好,背景压力低且塔板数较多,柱效较高,可在30 min 内实现7 种常规阴离子的分离,并且重现性和稳定性良好。本研究为制备聚附型阴离子固定相的产业化提供了良好的技术参考。
