色谱-串联质谱法测定天麻中53种禁用农药残留
2023-11-09罗京京程庆兵金凤华王玮
罗京京,程庆兵,金凤华,王玮
(1.安徽省药品审评查验中心,合肥 230051; 2.合肥市食品药品检验中心,合肥 230088)
天麻为兰科植物天麻的干燥块茎,是一种应用广泛的传统中药材,在历代本草文献中均有记载。天麻具有息风止痉、平抑肝阳、祛风通络之作用,临床用于小儿惊风、癫痫抽搐、破伤风、头痛眩晕、手足不遂、肢体麻木、风湿痹痛[1-2]。因需求量急剧上升,野生天麻资源濒临枯竭,目前主要以人工栽培为主,栽培地集中在贵州、安徽、云南、陕西、湖北等地[3-4]。
现阶段,中药材种植过程中使用农药现象较普遍。为保证中药质量,保障人民健康,应严格控制农药、化肥等在中药材种植环节中的使用。《中国药典》2020年版首次提出禁用农药残留量检测要求,对高毒、高风险农药严格把控,从源头管控中药质量[5-6]。近年来,天麻农药残留超标情况时有发生,对临床用药带来较大风险。目前,农药残留的检测方法主要有气相色谱法[7-8]、液相色谱-串联质谱法[9-12]、气相色谱-串联质谱法[13-15]等。胡德禹等[7]采用气相色谱法测定了天麻中有机氯农药残留;郭汉文等[8]采用气相色谱法测定了天麻中12有机氯农药残留;张婷等[13]采用气相色谱-质谱法测定了天麻中二硫代氨基甲酸酯类农药残留;冯红等[16]采用色谱-串联质谱法测定三七中禁用农药残留;杨红等[9]采用色谱-串联质谱法测定丹参中禁用农药残留;周琴等[10]采用色谱-串联质谱法测了前胡中禁用农药残留。采用色谱-串联质谱法测定天麻中53种禁用农药残留的方法尚未见报道。笔者参照文献[11]、[15]、[16],采用乙腈直接提取法和基于QuEChERS 原则的固相萃取法对天麻样品进行处理,用超高效液相色谱-串联质谱法及气相色谱-串联质谱法联合测定天麻中53 种禁用农药残留量。该方法操作简便,灵敏度高,可分析天麻禁用农药残留量情况,为市场上天麻的质量评价提供数据支撑。
1 实验部分
1.1 主要仪器与试剂
超高效液相色谱-三重四极杆质谱联用仪:ACQUITY XEVO TQ-S 型,美国沃特世科技有限公司。
气相色谱-三重四极杆串联质谱联用仪:8890-G7000D型,美国安捷伦科技有限公司。
电子天平:XPE205 型,感量为0.01 mg,瑞士梅特勒-托利多公司。
旋涡振荡器:VORTEX3型,德国艾卡公司。
离心机:Centrifuge 5417R 型,德国Eppendorf公司。
氮吹仪:MV5型,北京莱伯泰科公司。
30种农药混合标准溶液:编号为1ST 29486-A,各组分质量浓度见表1,天津阿尔塔科技有限公司。

表1 30种农药混合标准溶液中各组分质量浓度μg/mL
33 种农药混合标准溶液:编号为81208a,含有α-六六六、γ-六六六、β-六六六、δ-六六六、4,4′-滴滴伊、2,4′-滴滴涕、治螟磷、特丁硫磷、氟虫腈亚砜、氟虫腈、对硫磷、甲基对硫磷、内吸磷、杀虫脒、久效磷、甲基异硫磷、蝇毒磷、4,4′-滴滴滴、4,4′-滴滴涕、β-硫丹、α-硫丹、硫丹硫酸酯、狄氏剂、三氯杀螨醇、氟虫腈砜、除草醚、甲基硫环磷、艾氏剂、氟甲腈、灭线磷、甲拌磷、水胺硫磷、苯线磷,各组分质量浓度均为100 μg/mL,北京坛墨质检科技有限公司。
磷酸三苯酯标准溶液:100 μg/mL,溶剂为甲苯,编号为BW 901609-100-N,北京坛墨质检科技有限公司。
乙腈:HPLC级,美国默克公司。
QuEChERS 试剂盒:含盐包和净化管,批号为22 A217PY2,北京凯思科科技公司。
天麻样品:某单位提供。
1.2 样品处理
1.2.1 乙腈直接提取法
称取过3#筛的天麻样品粉末5.0 g,置于离心管中,加入1 g 氯化钠,摇散;再加入50 mL 乙腈,在旋涡振荡器上高速旋涡2 min,充分混匀;再于4 ℃下以4 000 r/min 转速离心5 min,分取上清液;向沉淀中再加入50 mL乙腈,匀浆处理1 min,离心;合并两次提取的上清液,减压浓缩至约5 mL,冷却,用乙腈稀释至10.0 mL,摇匀,待测。
1.2.2 基于QuEChERS原则的处理方法
称取过3#筛的天麻样品粉末3.0 g,置于50 mL聚苯乙烯具塞离心管中,加入10 mL 水及质量浓度为0.01 g/mL 的冰乙酸溶液15 mL,涡旋,使药粉充分浸润,放置30 min;加入乙腈15.00 mL,涡旋,混匀,以500 次每分钟的速率剧烈震荡5 min,加入QuEChERS盐包,立即摇散,再以500次每分钟的速率剧烈震荡3 min,于冰浴中冷却10 min,于4 ℃下以4 000 r/min 离心5 min,取上清液9 mL,置于QuEChERS净化管中,涡旋使其充分混匀,以500次每分钟的速率剧烈震荡5 min,使之净化完全,于4 ℃下以4 000 r/min 离心5 min,精密吸取上清液5 mL,置于氮吹仪上,于40 ℃水浴中浓缩至约0.4 mL,用乙腈稀释至1.0 mL,涡旋,混匀,待测。
1.3 液相色谱-质谱法仪器工作条件
1.3.1 液相色谱仪
色谱柱:ACQUITY UPLC BEH C18柱[100 mm×2.1 mm,1.7 μm,沃特世科技(上海)有限公司];流动相:A 相为0.1%(体积分数,下同)甲酸溶液,B 相为乙腈-0.1%甲酸溶液(体积比为95∶5),A、B 相均含有5 mmol/L 甲酸铵;洗脱方式:梯度淋洗,0~1 min时70% A,1~12 min 时70%~0% A,12~14 min 时0%~100% A;流动相流量:0.3 mL/min;柱温:40 ℃;进样体积:1 μL。
1.3.2 质谱仪
离子模式:电喷雾电离,正离子扫描;毛细管电压:2.50 kV;离子源温度:150 ℃;脱溶剂气温度:500 ℃;锥孔气:氮气,流量为20 L/h;脱溶剂气:氮气,流量为1 000 L/h。29种农药化合物质谱参数见表2。

表2 29种农药化合物质谱参数
1.4 气相色谱-质谱法仪器工作条件
1.41 气相色谱仪
色谱柱:DB-17MS 弹性石英毛细管柱(30 m×0.25 mm,0.25 μm,美国安捷伦科技有限公司);进样口温度:250 ℃;进样方式:不分流;进样体积:1 μL;柱箱温度:程序升温,初始温度为60 ℃,保持1 min,以30 ℃/min的速率升温至120 ℃,然后以10 ℃/min的速率升温至160 ℃,再以2 ℃/min 的速率升温至230 ℃,最后以15 ℃/min的速率升温至300 ℃,保持6 min;载气:高纯氦气,流量为1.0 mL/min。
1.4.2 质谱仪
电离方式:电子轰击电离模式;电离能量:70 eV;离子源温度:250 ℃;质谱传输接口温度:250 ℃;碰撞气:高纯氮气;四级杆温度:150 ℃;扫描方式:多反应监测模式(MRM)。24种农药化合物质谱参数见表3。

表3 24种农药化合物质谱参数
1.5 溶液配制
29 种农药(具体名称见表2)混合标准储备液:精密量取30 种农药混合标准溶液1 mL,置于100 mL容量瓶中,用乙腈稀释并定容至标线,摇匀。
基质匹配系列混合标准工作溶液A:取天麻空白基质样品,按1.2.1方法处理。取样品提取液6份,各1.0 mL,分别于40 ℃水浴中浓缩至约0.6 mL,分别加入29 种农药混合标准储备液10、20、50、100、150、200 µL,用乙腈稀释至1.0 mL,涡旋,混匀。
24种农药(具体名称见表3)混合标准储备液:1 μg/mL,精密量取33 种农药混合标准溶液0.5 mL,置于50 mL容量瓶中,用乙腈定容至标线,摇匀。
磷酸三苯酯溶液:1 ng/mL,精密量取磷酸三苯酯标准溶液50 µL,置于50 mL 容量瓶中,用乙腈定容至标线,摇匀。精密量取上述溶液1 mL,置于100 mL容量瓶中,加乙腈稀释并定容至标线,摇匀。
基质匹配系列混合标准工作溶液B:取天麻空白基质样品,按1.2.2方法处理。取样品提取液6份,各1.0 mL,分别于40 ℃水浴中浓缩至约0.6 mL,分别加入24种农药混合标准储备液5、10、20、50、100、150、200 µL,用乙腈稀释至1.0 mL,涡旋,混匀。
1.6 定量方法
1.6.1 液相色谱-质谱法
取处理好的样品溶液和基质匹配系列混合标准工作溶液A各1 mL,分别精密加入0.3 mL水,混匀,滤过,取续滤液进样测定,记录色谱图,以色谱峰面积标准曲线法定量。
1.6.2 气相色谱-质谱法
取处理好的样品溶液和基质匹配系列混合标准工作溶液B各1 mL,分别精密加入0.3 mL磷酸三苯酯溶液作为内标,混匀,滤过,取续滤液进样测定,以标准曲线内标法定量。
2 结果与讨论
2.1 质谱条件优化
2.1.1 液相色谱-质谱法质谱条件
分别选择在ESI 正离子和负离子模式下2 种扫描方式,对29 种目标农药化合物进行一级质谱扫描,获得稳定的母离子分子离子峰,然后对其进行二级扫描,获得二级离子质谱信息,筛选2个信号较强的离子碎片作为子离子,并在MRM 模式下分别优化锥孔电压、碰撞能量等参数,选取响应强度高且基质干扰小的两对离子作为定量和定性离子,优化结果见表2。
2.1.2 气相色谱-质谱法质谱条件
对24种目标农药化合物进行扫描,选择有代表性的母离子,选取特征离子作为子离子,建立多反应监测模式,同时优化定量、定性离子对的碰撞能量,优化结果见表3。空白样品和加标样品色谱图如图1所示。
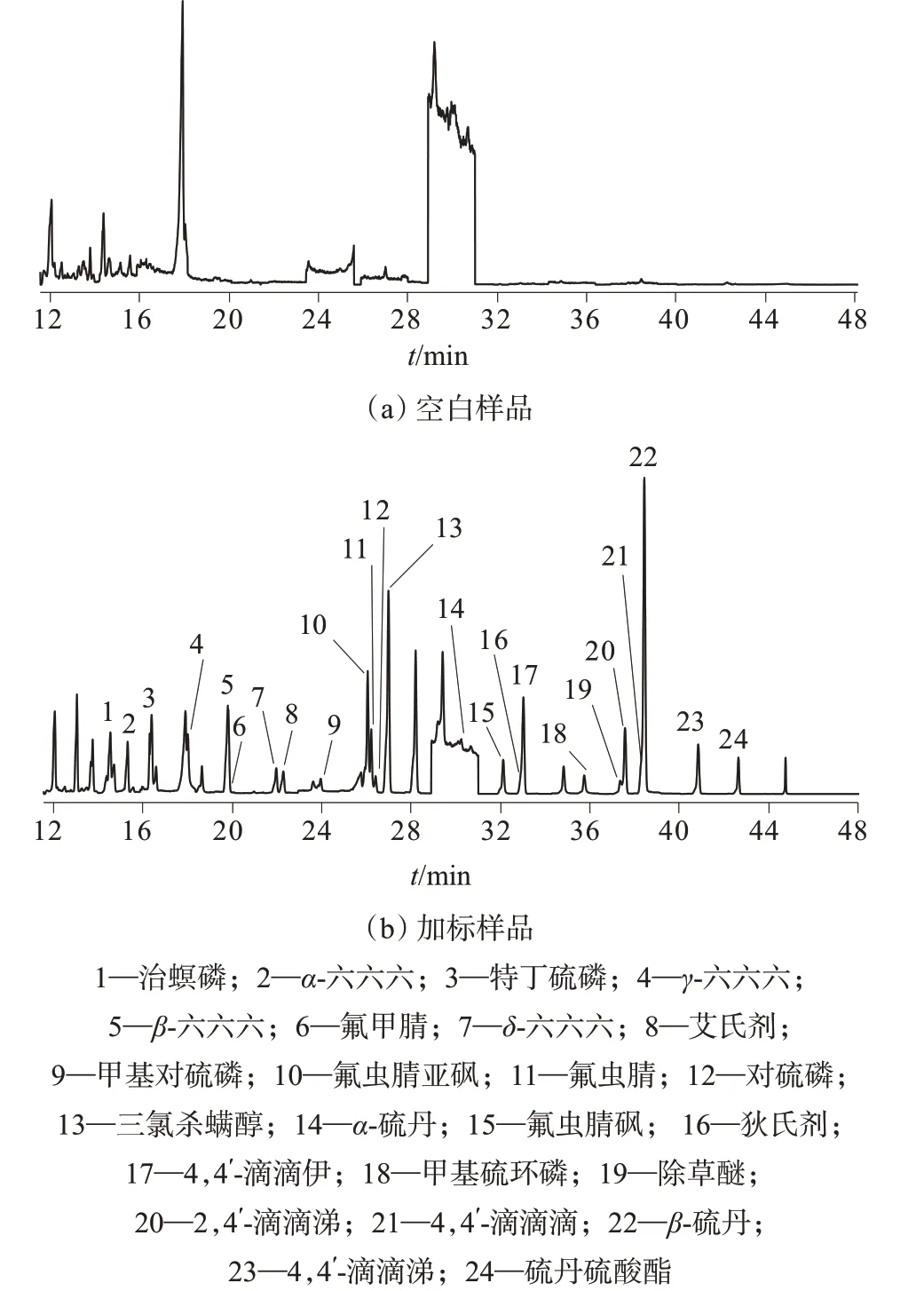
图1 空白样品及加标样品气相色谱图
2.2 样品处理方法优化
2.2.1 基质效应
以农药化合物在基质匹配标准溶液中色谱峰面积与其溶剂标准溶液中的信号峰面积比值计算基质效应,若比值大于120%,认为存在强基质效应;若比值为80%~120%,则认为没有基质效应;若比值小于80%,则认为存在弱基质效应。
分别采用乙腈直接提取法和基于QuEChERS原则的固相萃取法处理样品,发现以乙腈直接提取法处理的样品,采用LC-MS/MS 法测定时,只有约20%的目标物存在基质效应,且基质效应较小;而采用GC-MS/MS 法测定时,基质效应较强,有近50%的目标物存在基质效应,但基质效应多数较弱。采用基于QuEChERS 原则的固相萃取法处理样品,以LC-MS/MS法测定时,有约17%的目标物存在基质效应,且基质效应较小;以GC-MS/MS法测定时,有约39%的目标物存在基质效应。综合考虑,选择采用乙腈直接提取法处理样品用于LC-MS/MS 法测定,采用基于QuEChERS 原则的固相萃取法处理样品用于GC-MS/MS法测定。
2.2.2 基于QuEChERS原则的固相萃取法
采用基于QuEChERS 原则的固相萃取法处理样品,发现部分含磷、氯农药化合物(如三氯杀螨醇、治螟磷等)受天麻基质影响,回收率较低,可能与根类药材含水量低、样品未充分溶胀有关,导致提取效率低。通过对样品中水的加入量进行比较,考察加水量分别为5、10、15 mL时农药化合物的回收率,结果表明,在加入水的体积小于10 mL时,增加水的用量,会显著提高含磷、氯农药化合物的回收率,当加入水的体积大于10 mL时,回收率不再明显增加,因此选择加水量为10 mL。
2.3 线性方程与检出限
在1.4、1.5仪器工作条件下,分别测定基质匹配系列混合标准工作溶液A、B,以各目标物的色谱峰面积对其质量浓度进行线性回归,计算线性方程和相关系数。
取基质匹配系列混合标准工作溶液A、B 的最低浓度点标准溶液,用乙腈逐级稀释,测定并记录色谱峰面积,以信噪比大于等于3 计算目标物在样品中的质量分数,作为方法检出限,以信噪比大于等于10计算方法定量限。
53 种农药的质量浓度线性范围、线性方程、相关系数、检出限、定量限见表4。由表4可知,53种目标物在一定质量浓度范围内,相关系数均不小于0.990,表明线性关系良好,方法检出限为0.001~0.005 g/kg,定量限为0.002~0.01 mg/kg,满足农药残留的分析要求。
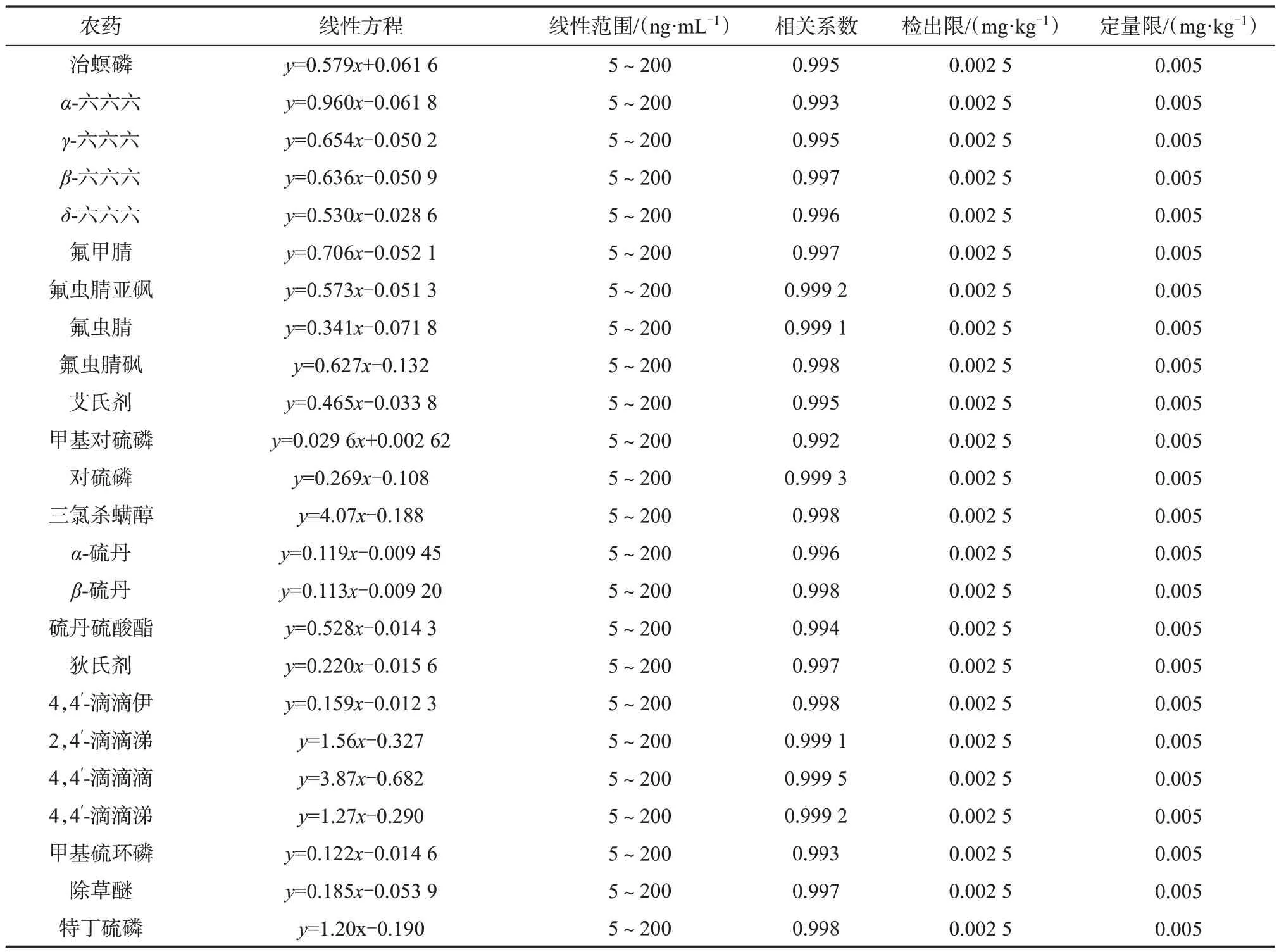
表4 53种农药质量浓度线性范围、线性方程、相关系数、检出限及定量限
2.4 加标回收与精密度试验
取空白基质样品,分别添加低、高两个浓度水平的混合标准溶液,平行测定6次,以平均值作为测定值,用基质匹配标准曲线法定量,结果见表5。由表5 可知,各目标物平均回收率为71.4%~118.2%,相对标准偏差为1.4%~6.3%(n=6),表明方法的回收率和重现率良好,符合禁用农药残留测定的要求,适用于天麻中53种农药残留的同时测定。

表5 加标回收与精密度试验结果(n=6)
3 结语
建立了液相色谱-串联质谱法和气相色谱-串联质谱法同时测定天麻中53 种禁用农药残留的方法。采用乙腈直接提取和基于QuEChERS 原则的固相萃取法对样品进行处理,以基质匹配标准工作曲线法定量,能快速实现天麻中禁用农药化合物的含量分析。该方法具有较好的准确度和精密度,满足复杂基质中痕量农药分析要求,且简单、快速、稳定,可用于批量天麻样品中禁用农药残留的快速分析测定。
