基于家系基因组测序对骡基因组结构多样性分析
2023-11-09任秀娟苏少锋李雅静蔺雅楠贾紫洁丁文淇白东义赵一萍
任秀娟 苏少锋 李雅静 蔺雅楠 贾紫洁 丁文淇 白东义 李 蓓 杜 明 芒 来 赵一萍*
(1.内蒙古农业大学 动物科学学院/马属动物研究中心,呼和浩特 010018;2.内蒙古自治区农牧业科学院,呼和浩特 010031)
马(Equuscaballus)和驴(Equusasinus)属于2个独立的物种,其物种分离发生在约400~450万年前[1]。马属动物的物种形成伴随着快速的染色体重排,其重排速率为2.9~22.2次/百万代[2-3]。马(2n=64)和驴(2n=62)已经进化形成了两套结构和功能完善的独立基因组[4-5]。生理特征比较表明,驴具有更有效的能量代谢和更强的免疫力,而马的反应更灵敏和运动能力更强[6]。马和驴杂交能够产生后代(马骡,2n=63,公驴和母马的杂交后代;驴骡,2n=63,公马和母驴的杂交后代)。尽管有母骡产驹的报道,但骡基本上不能自然交配产生后代。
根据Dobzhansky-Muller不相容定律(Dobzhansky-Muller incompatibility),马和驴物种间进化分离的2个或多个等位基因之间不能有效的相互作用,可能导致骡的适应性存在缺陷,表现为不育和溶血等[7-8]。例如,物种形成基因的反复适应性进化[9]、细胞质基因和核基因的冲突进化[10]以及X染色体的减速分裂驱动等[11],均可导致异种杂交不相容。剧烈的染色体结构变异也可能是马和驴杂交不相容的遗传因素。例如,染色体易位可导致部分异种杂交个体的某些基因完全丢失,引起不相容[12]。Prdm9基因的拷贝数差异,导致小鼠(Musm.musculus×Musm.domesticus)品种间雄性杂交个体减数分裂重组失败[13]。马和驴属于古老的物种,除了物种形成基因外,在长期的进化过程中,其基因组之间累积了更复杂的遗传不相容。例如,受平衡选择形成种间多态性的免疫相关基因,是许多物种间杂交障碍的主要原因。对小鼠[14]和硬骨鱼[15]的研究表明,主要组织相容性复合物(Major histocompatibility complex,MHC)基因的种间多态性会降低F1杂交个体的适应性。
骡如何协调双亲基因组的不平衡并保证其自身生存的分子机制仍然未知。大量研究表明,在胚胎发育早期,会发生合子后体细胞突变,同时异常的染色体结构变异会触发机体的自我纠错过程。这些突变大部分会导致机体发生癌症和一些罕见的发育障碍疾病[16-17]。然而,大部分存活的杂交个体伴随着基因组的高频突变。例如,对植物的研究表明,高度杂合个体的全基因组突变率高于纯合个体[18-19]。异种杂交可能会激活大量转座子的活性,从而促使更多结构变异的形成。例如,金鱼(Carassiusauratus)和鲤鱼(Cyprinuscarpio)杂交,其F1杂交个体基因组发生了高频率的点突变和大片段的结构变异[20]。王艳欣[21]对马骡和驴骡的膀胱等10个组织转录组的研究发现,骡不同组织均发生了高频率的结构变异。
异种杂交个体发生的高频突变,可能反映了物种间进化累积的差异突变导致的遗传不相容。但是,杂交应激触发的快速突变,是否能缓冲其亲本单倍体基因组之间的不平衡,从而提高杂交个体的适应性尚不清晰。本研究基于马属动物三成员家系的全基因组Illumina测序数据,旨在分析马和驴杂交应激触发的骡基因组结构多样性,揭示马和驴遗传不相容以及影响骡适应性的可能基因座,为进一步开展马和驴异种杂交的遗传基础及其分子机制的研究提供候选遗传位点。
1 材料与方法
1.1 实验动物和测序数据
本研究的实验动物包括1匹雌性蒙古马、1匹雄性家驴和1匹雌性马骡。3匹实验动物来自于1个马属动物异种杂交家系。基因组序列由内蒙古农业大学马属动物研究中心产生和存储。所有数据已提交NCBI,驴的项目登录号:PRJNA205517[6],马和骡的项目登录号:PRJNA842856(SRA登录号:SRR19427107和SRR19427108)[22]。
1.2 驴、马和骡参考基因组的序列比对
以纯血马基因组(Equcab3.0)[5]和驴基因组(ASM1607732v2)[23]为参考,使用BWA(version 0.7.5a-r416)软件的默认参数,将驴、马和骡的高质量reads比对到参考基因组[24]。使用SAMtools(version 0.1.19-44428cd)软件获得唯一比对,参数为“-q 30”[25]。使用Picard(version 1.93,http:∥sourceforge.net/projects/picard/)软件标记测序过程产生的潜在重复。
1.3 驴、马和骡SNP和InDel的识别
使用SAMtools和GATK(version 3.5-0-g36282e4)软件[26]对3个样本分别识别SNP和InDel。2个软件分析结果的交集用于后续分析。使用GATK的HaplotyCaller模块识别SNP,参数为-standa_call_conf 30,-standa_emit_conf 10。采用VariantFiltration命令,根据(http:∥www.broadinstitute.org/gatk/guide/best-practices)推荐参数对获得的SNP进行严格过滤。为提高SNP的阳性率,在上述严格过滤的基础上,进一步过滤掉符合以下4个标准的SNP:(i)位于低复杂度或简单重复区域;(ii)测序深度<4或>50;(iii)InDel周围50 bp范围内;(iv)Gap周围10 bp范围内,至少3个SNPs被识别。
1.4 骡de novo SNP的识别
骡denovoSNP是指针对参考基因组的一个碱基位点,骡基因组的该位点至少有1个等位基因不同于亲本。骡denovoSNP基因型组合见表1。采用两种方法分别识别denovoSNP,并取交集用于后续分析。首先,参考Roach的方法[27],利用bcftools软件的query工具和vcftools软件,按表1的基因型组合,识别骡denovoSNP。其次,使用VarScan软件的“trio”命令,识别骡denovoSNP,参数:--min-coverage 10,--min-var-freq 0.20,--p-value 0.05,--adj-var-freq 0.05,--adj-p-value 0.15[28]。将两种方法获得的denovoSNP取交集。本研究三成员家系的试验设计,因为基因型的错误分配,可能会影响骡denovoSNP的准确识别[29]。因此,将定位在拷贝数变异(CNV)区域和重复序列区域的SNP屏蔽掉。用CNVnator软件[30]检测CNV和用RepeatMasker软件[31]检测重复序列。用于后续分析的denovoSNP符合以下2个标准:1)SNP位点的reads支持数≥10;2)SNP等位位点的reads支持数≥5[32]。
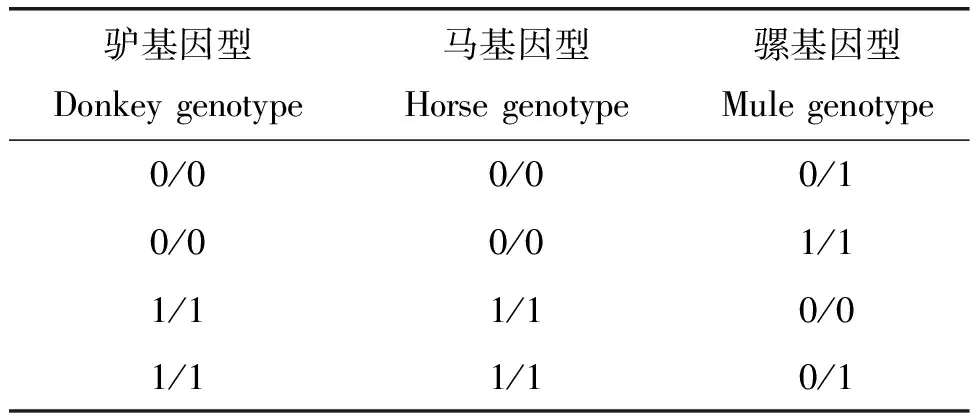
表1 De novo SNPs的基因型组合
ANNOVAR软件用于denovoSNP的功能注释。对于定位到基因间区的SNP,仅保留上下游5 kb范围内的基因。
1.5 驴、马和骡CNV检测及注释
使用CNVnator软件及其推荐参数识别CNV[30]。使用以下标准过滤原始CNV:获得性CNV(duplication)的RD(normalized read depth)>2,缺失性CNV(deletion)的RD<0.4,P≤0.05,q0≤0.5,Length>1 000 bp。将驴、马和骡位置重叠≥1 bp的CNV,合并为1个拷贝数变异区域(CNVR)。
使用bedtools软件对CNV进行基因注释,标准是CNV的bed文件和基因组的gtf文件至少有1个碱基的重叠[33]。
1.6 基因富集分析
使用R软件的clusterProfiler包,进行KEGG富集分析,纯血马参考基因组的参数设置为:organic=“ecb”,keyType=“KEGG”;驴参考基因组的参数设置为:organic=“eai”,keyType=“KEGG”。
2 结果与分析
2.1 驴、马和骡测序数据及比对
经严格质控后存储于内蒙古农业大学马属动物研究中心的驴、马和骡的高质量基因组数据,分别为100.01、103.78和114.36 Gb[6,22]。将基因组数据分别比对到纯血马参考基因组(Equcab3.0)和驴参考基因组(ASM1607732v2)。以纯血马基因组为参考和以驴基因组为参考时,至少98%的基因组覆盖深度大于1×。从表2可知,分别以马和驴的基因组为参考,读段的比对率不存在比对的偏向性。
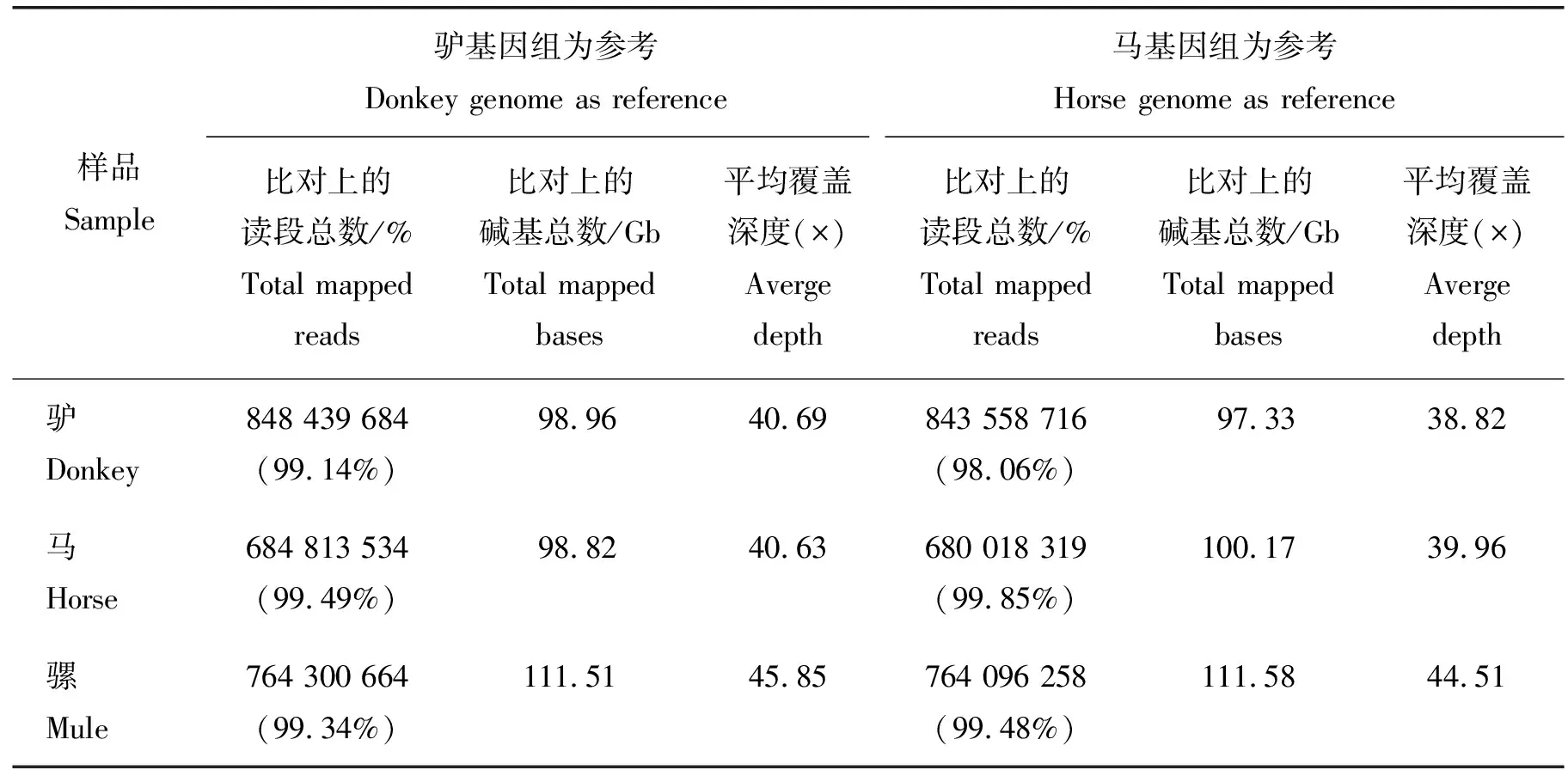
表2 参考基因组比对结果
2.2 驴、马和骡SNP和InDel检测分析
2.2.1驴、马和骡InDel和SNP识别
以纯血马基因组为参考,驴、马和骡识别的高质量InDels分别为2 110 786、402 533和1 816 910。以驴基因组为参考,驴、马和骡识别的高质量InDels分别为527 351、2 279 875和2 036 942。以纯血马基因组为参考时,驴和骡的InDel数明显高于马,这符合蒙古马和纯血马的亲缘关系更近,骡是驴和马的杂交个体。同样的原因,当以驴基因组为参考时,马和骡的InDel数明显高于驴。由图1可知,InDel基本均匀地分布于常染色体。
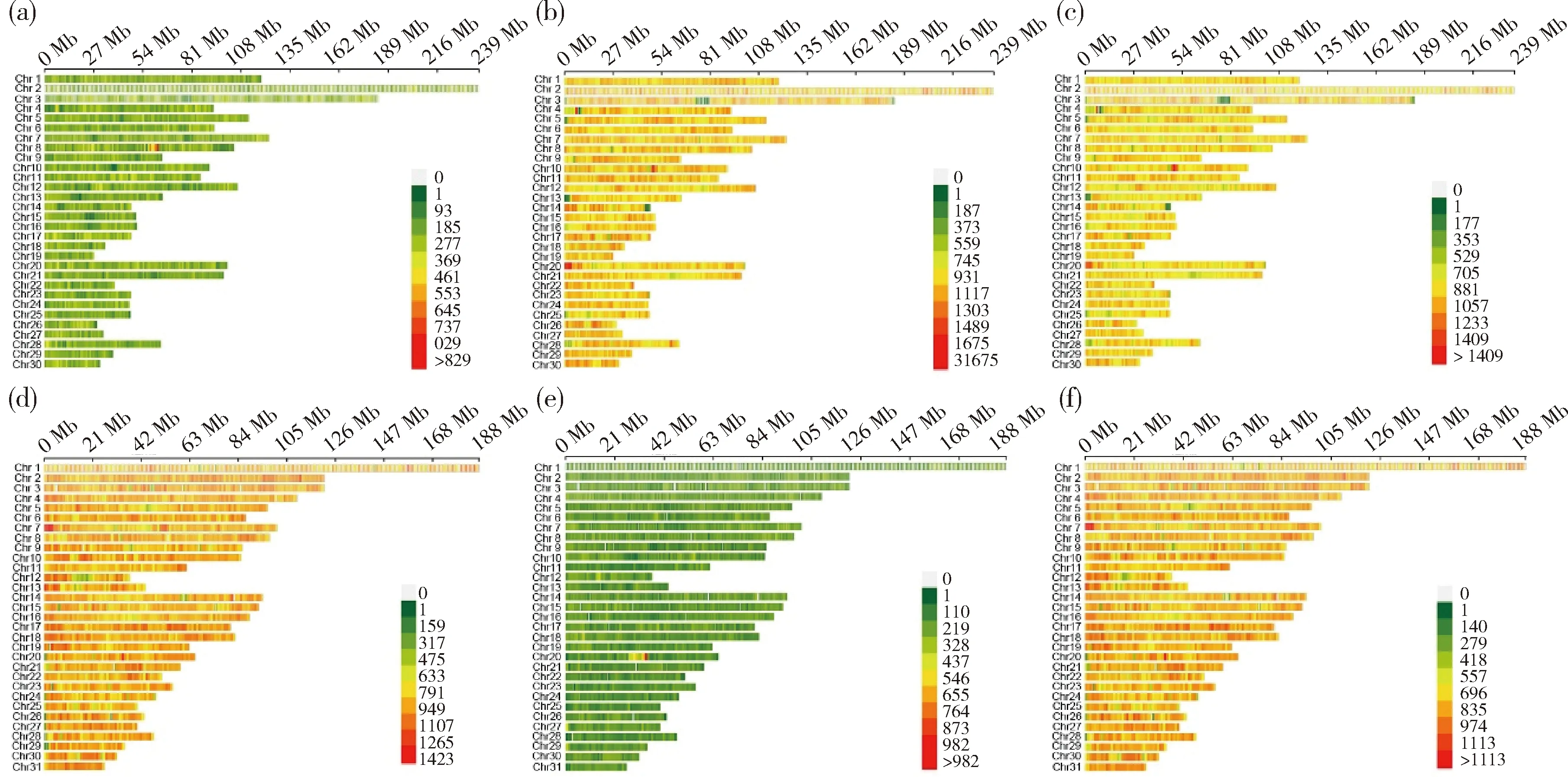
(a)、(b)和(c)表示以驴基因组为参考时InDel的常染色体密度分布。(d)、(e)和(f)表示以纯血马基因组为参考时InDel的常染色体密度分布。(a)和(d)表示驴;(b)和(e)表示马;(c)和(f)表示骡。
以驴基因组为参考,分别识别驴、马和骡的高质量SNPs为3 212 499、23 549 224和21 870 390。马(0.968 3%)和骡(0.899 3%)的SNP频率明显高于驴(0.132 1%)。实验室已发表数据表明,以纯血马基因组为参考,分别识别驴、马和骡的高质量SNPs为23 819 055、5 012 403和23 426 241;蒙古马共识别5 012 403个SNPs,杂合SNP数高于纯合,驴(0.950 1%)和骡(0.934 4%)的SNP频率明显高于马(0.199 9%)[22]。以上结果表明,由于驴、马和骡与参考基因组物种的亲缘关系不同,导致与参考基因组不同物种识别的SNPs频率远高于相同物种(表3)。以纯血马基因组为参考识别的骡SNPs数略高于以驴基因组为参考的SNPs识别结果。由图2可知,SNP基本均匀分布于常染色体。
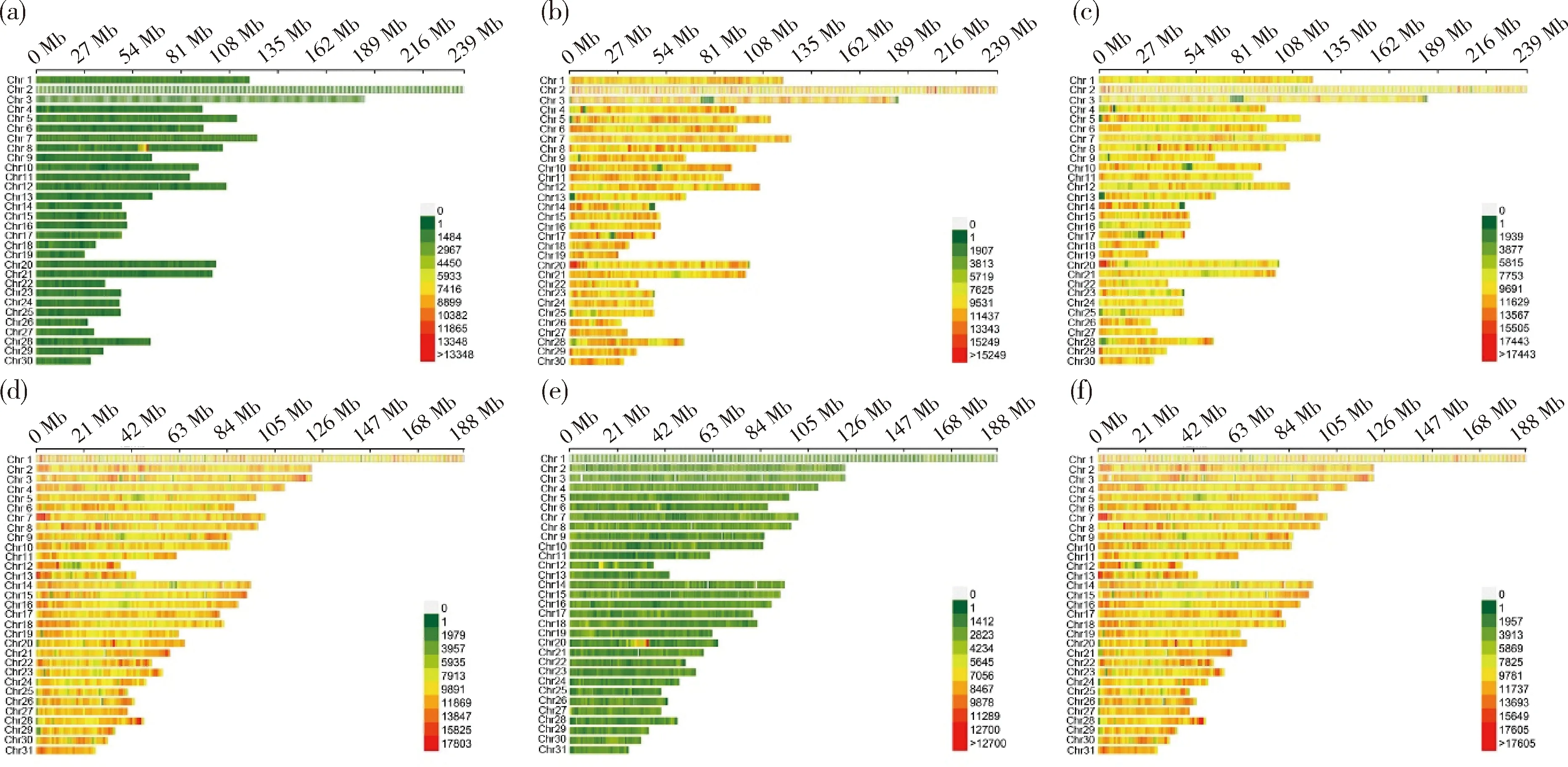
(a)、(b)和(c)表示以驴基因组为参考时SNP的常染色体密度分布。(d)、(e)和(f)表示以纯血马基因组为参考时,SNP的染色体密度分布[22]。(a)和(d)表示驴;(b)和(e)表示马;(c)和(f)表示骡。
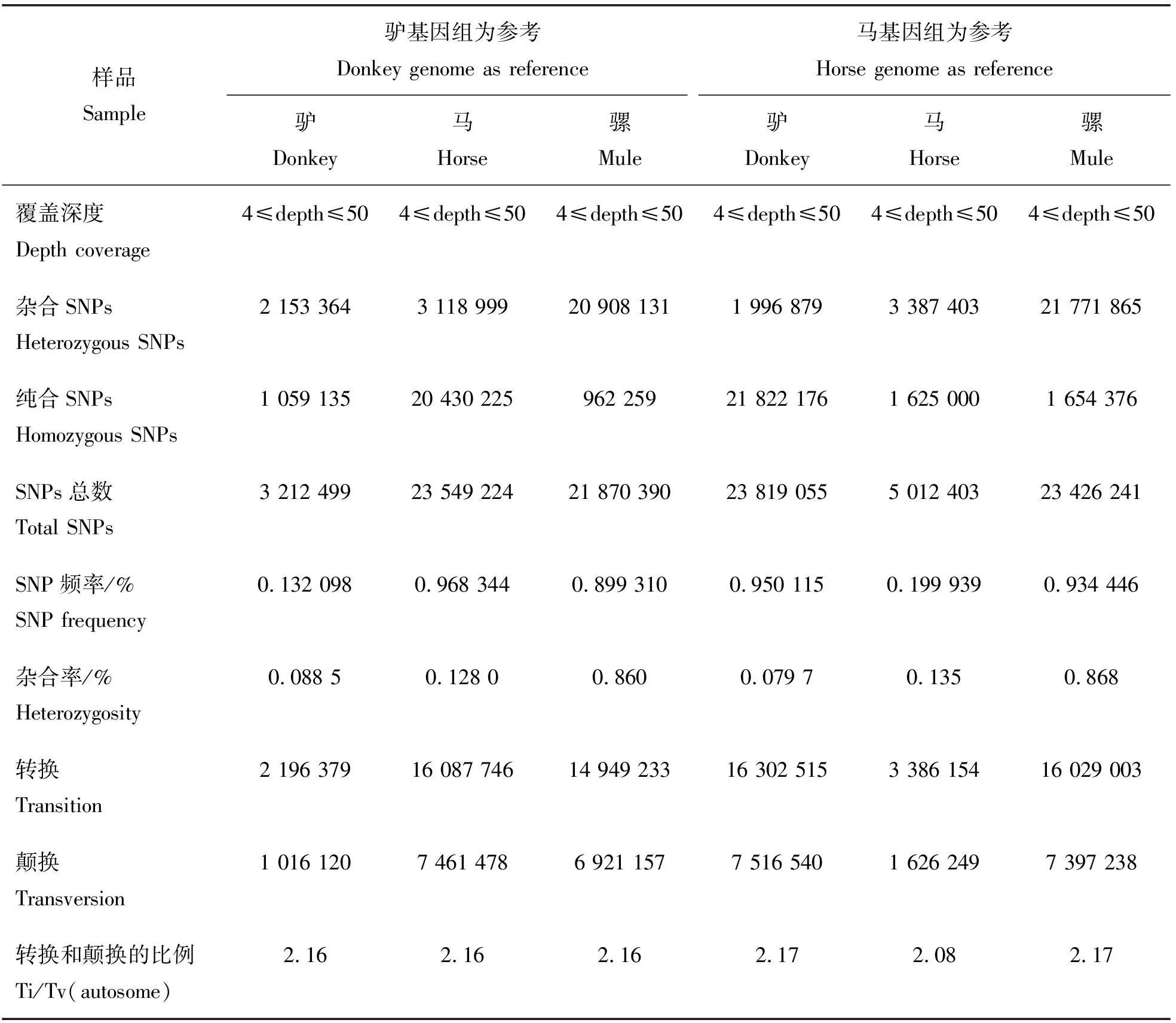
表3 SNPs的统计
2.2.2骡denovoSNP
骡denovoSNP是指针对参考基因组的1个碱基位点,骡的该位点至少有1个等位基因不同于亲本。对上述识别的高质量SNP,进一步屏蔽掉重复序列和CNV区域的SNP。以纯血马基因组和驴基因组分别为参考,与亲本相比,骡分别识别了555和419个denovoSNPs。与SNP识别趋势一致,骡denovoSNP的识别可能存在马参考基因组的偏向性。本研究的三成员家系中,骡denovoSNP的频率是1.72×10-7~2.21×10-7(DenovoSNPs/全基因组序列)。
2.2.3骡denovoSNP相关基因注释
为研究骡denovoSNP的潜在功能,使用ANNOVAR软件,对SNP上下游5 kb范围内的基因进行注释。以纯血马基因组为参考,555个denovoSNPs共注释658个相关基因。以驴基因组为参考,419个denovoSNPs共注释540个相关基因。DenovoSNP均最高频率的注释于基因间区(表4)。以两套基因组为参考注释denovoSNP,均发现大部分相关基因直接参与机体的免疫功能。例如,纯血马参考基因组注释的基因,包括MHC I类基因(如MHCX1和LOC100049798)和MHC II类基因(如DQA和DQB)、Src家族酪氨酸激酶(如FYN)、表皮生长因子受体基因(EGFR)以及癌症相关基因(如APC和PTEN、HRAS)[22]。驴参考基因组注释的基因,包括MHC I类基因(如LOC106825028和LOC106843523)和MHC II类基因(如LOC106830318和LOC106834623)、Src家族酪氨酸激酶(如FYN)、表皮生长因子受体基因(EGFR)以及肿瘤抑制基因(如APC和PTEN)。
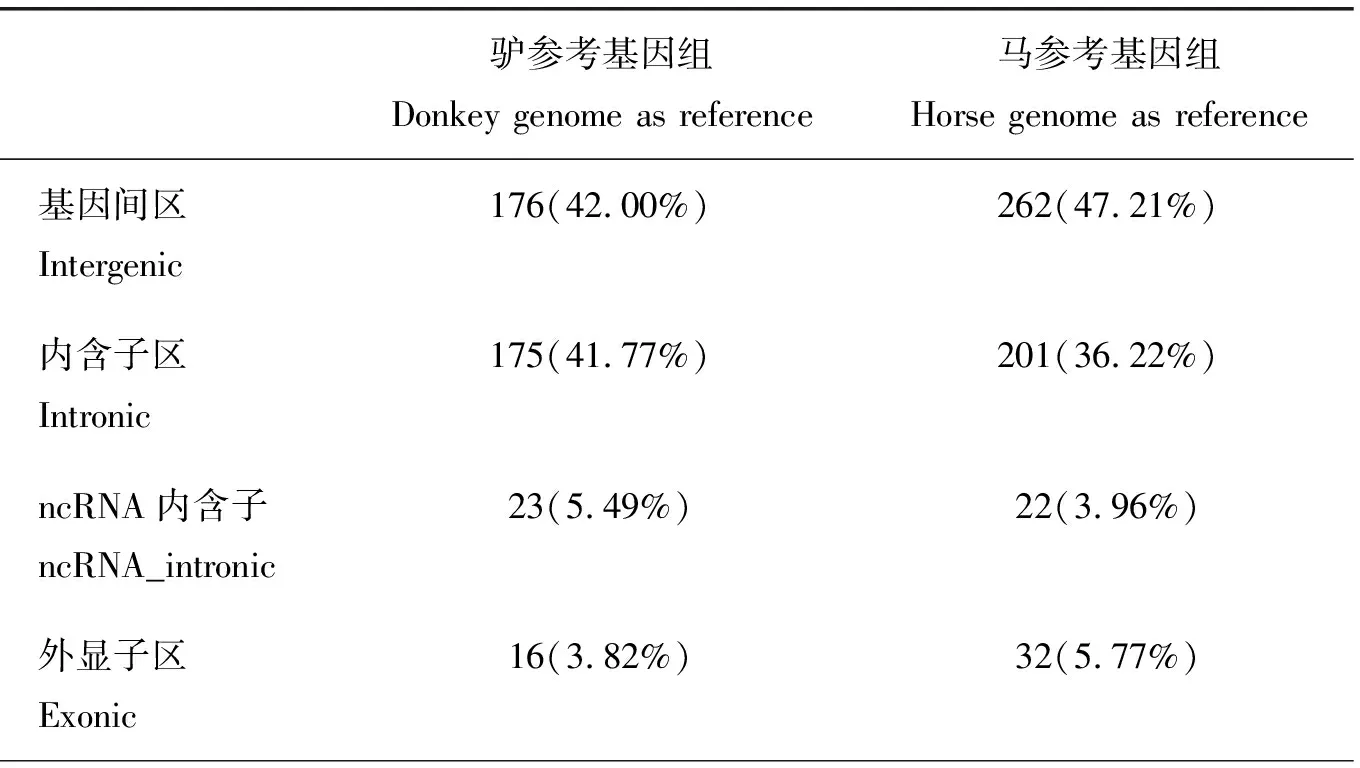
表4 De novo SNP的分布统计
2.2.4骡denovoSNP相关基因富集分析
对骡denovoSNP相关基因进行KEGG富集分析,发现大部分显著富集(P<0.05)的通路参与机体的免疫应答过程。以纯血马基因组和驴基因组为参考时,分别有43和27条通路被显著富集,其中17条通路是相同的(图3)。这些通路主要与机体的免疫过程相关,包括在适应性免疫应答中起关键作用的抗原加工和提呈过程;机体排异相关过程:移植物抗宿主病、同种异体移植排斥;以及自身免疫性疾病通路:I型糖尿病、类风湿性关节炎。

图3 De novo SNP相关基因的KEGG富集
2.3 驴、马和骡CNV特征分析
2.3.1驴、马和骡CNV的检测
以纯血马基因组为参考,驴、马和骡分别获得3 761、2 126和2 242个 CNVs(表5)。将CNVs合并后,共获得5 178个CNVRs(表6)。骡特异性CNVRs总长度为2.15 Mb,平均长度为5.43 kb。马和骡CNVRs总长度为7.64 Mb,平均长度为11.35 kb。驴和骡CNVRs总长度为5.41 Mb,平均长度为16.90 kb。驴、马和骡CNVRs总长度为13.05 Mb,平均长度为26.74 kb。
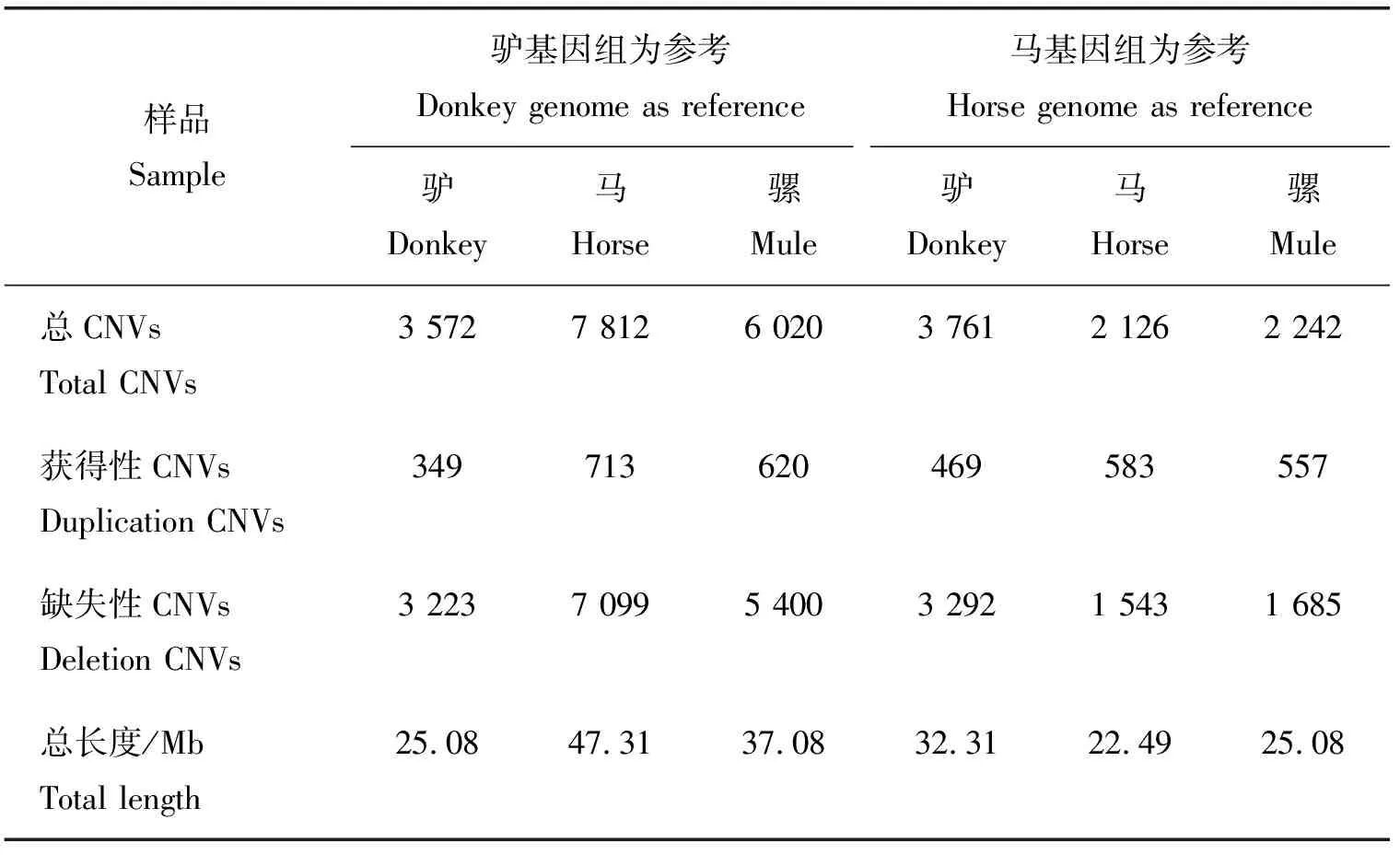
表5 CNVs的统计
以驴基因组为参考,驴、马和骡分别获得3 572、7 812和6 020个CNVs(表5)。将CNVs合并后,共获得8 967个CNVRs(表6)。骡特异性CNVRs总长度为3.77 Mb,平均长度为4.50 kb。马和骡CNVRs总长度为15.14 Mb,平均长度为7.93 kb。驴和骡CNVRs总长度为3.00 Mb,平均长度为7.79 kb。驴、马和骡CNVRs总长度为17.09 Mb,平均长度为11.39 kb。
2.3.2骡特异性CNVR相关基因功能分析
骡特异性CNVR是指相对参考基因组的某片段,马和驴无拷贝数变异且只有骡发生的CNVR。以纯血马基因组为参考时,396个骡特异性CNVRs,共注释226个基因。以驴基因组为参考,839个骡特异性CNVRs,共注释435个基因。
KEGG富集分析显示,以纯血马基因组为参考和以驴基因组为参考分别富集了93和145条pathways,其中共同富集的通路有66条。显著富集(P<0.05)的通路分别是42和13条。尽管一些通路的富集没达到显著水平,但是对于分析突变基因的相关功能仍然有意义。共同富集的66条pathways主要与机体的免疫过程相关(图4),包括抗原加工和提呈(如EQMHCB2和LOC100049798)、MAPK信号通路(如MAP3K2)、JAK-STAT信号通路(如AOX1)、NF-κB信号通路(如CARD14和TRIM25)、cGMP-PKG信号通路(如PDE5A和SLC8A2)。癌症相关过程,包括癌症途径(如APC2和SOS2)、Ras信号通路(如RAP1B)、Wnt信号通路(如NKD2)和癌症中转录调控异常等。
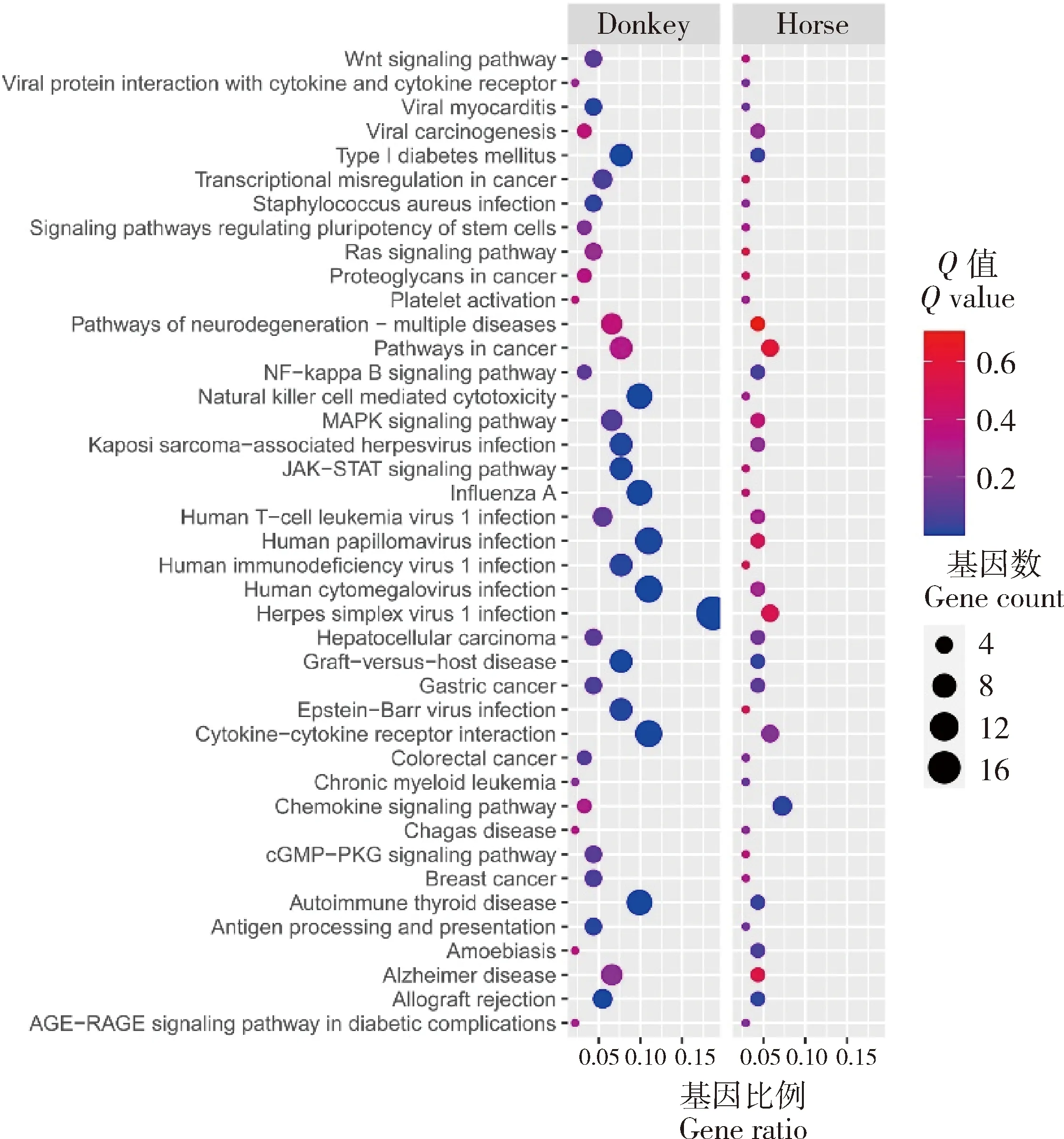
Donkey表示以驴基因组为参考时的富集结果;Horse表示以马基因组为参考时的富集结果。
3 讨 论
本团队已有研究表明,以纯血马基因组为参考,使用标准流程识别的驴和蒙古马SNPs及其频率与已有报道相符[22,34-36]。使用相同的方法,以驴基因组为参考,识别了驴2 153 364个杂合SNPs,该数值和已发表的研究数据(2 187 070,以驴Maral har基因组为参考)相近[6]。驴杂合SNP频率(0.088 5%)略高于Wang等[23]的报道(0.077 98%)。以上结果说明,本研究识别的SNPs数据可信。以驴基因组为参考识别的骡SNPs和denovoSNPs均略低于以纯血马基因组为参考的结果,说明SNP的识别可能存在纯血马参考基因组的偏向性。
结合以纯血马基因组为参考识别骡denovoSNPs(555)的结果可知[22],骡denovoSNPs数远高于用相同方法识别的纯种黑猩猩的denovoSNPs数(45)[37]。骡denovoSNP的突变率(1.72×10-7~2.21×10-7)高于人类(0.82×10-8~1.70×10-8)[38-39]和马(7.24×10-9)[1]的自然突变率。骡基因组更高的突变频率,符合Duncan在1915年通过实验验证的假设,即异种杂交个体具有高的突变率[40]。根据已有的文献报道[39,41-42],推测骡基因组中较高的denovoSNP频率,是由于马和驴单倍体整合到骡基因组的应激引发的合子后快速突变所致。但本研究的数据不足以解释其详细分子机制。
研究表明,用相同的方法识别不同品种马的CNV,其数量变化范围为从几十到几千[43]。而不同的方法对CNV识别的敏感度也有很大差别[44]。CNVnator软件对CNV的识别具有高灵敏度(86%~96%),用该软件对6个人深度测序(20×~32×)的illumina数据进行CNV分析,共识别了737~1 489个deletion CNVs[30]。使用CNVnator软件,本研究识别的马和驴deletion CNVs分别是1 543和3 223。与2014年Wang等[45]和2012年Doan等[46]的研究结果相比,本研究马和驴CNVs数量相对较高,这可能与使用的分析方法、数据类型以及品种等有关。以驴基因组为参考识别的CNVs数高于以纯血马基因组为参考,该结果与韩红梅[47]的研究结果趋势一致,但与之相比本研究识别的骡CNVs数量更少,这可能是因为本研究对CNV的过滤更严格。与SNP相比,CNV结构变异对机体具有更大的影响。马和驴物种间的CNV以及相关基因间的遗传距离,导致评估CNV对骡的影响非常复杂。
功能分析发现许多变异相关基因和机体的免疫过程相关。例如MHC基因家族,MHC基因是机体维持免疫稳态和发挥适当免疫功能的关键基因。与大多数哺乳动物一样,马属动物MHC基因的特征是由病原微生物平衡选择维持的极端多态性,和由不同地理环境导致的抗原结合位点的谱系特异性[48-50]。Masly等[12]对异种杂交小鼠的研究结果表明,MHC基因的种间多态性会降低其杂交后代的适应性、存活率或繁殖成功率。许多在免疫过程中发挥关键作用的通路,在本研究中被富集,包括抗原加工和提呈过程、TCR信号传递的2个重要下游信号通路:MAPK信号通路和NF-κB信号通路[51]、以及在T细胞分化过程中具有重要调控作用的JAK-STAT信号通路[52]。王艳欣[21]对马骡和驴骡不同组织转录组的研究发现,许多结构变异相关基因富集到了免疫相关通路。韩红梅[47]对马骡和驴骡基因组的研究发现,许多结构变异相关基因和机体免疫过程相关。骡部分结构变异相关基因还参与机体的癌症过程,例如,原癌基因(如HRAS)和肿瘤抑制基因(如APC和PTEN)的突变是各种癌症发生的主要候选基因[53-55]。Liu等[20]对杂交鱼的研究发现,许多结构变异相关基因和癌症过程相关。综上所述,这些突变可能反映了马和驴之间进化累积的不相容位点,或者是反映了马和驴杂交在基因组序列水平是不利于骡适应性的[7]。另一方面,突变产生的结构变异,可能通过改变酶活性或专一性,影响基因的正常转录及最终的功能效能[56]。因此,快速突变的SNP也可能作为一种“缓冲剂”来平衡亲本单倍体基因组的不相容,从而提高骡的适应性[7]。因此,在接下来的工作中,需要在更大样本量、更高测序深度和更多试验开展的基础上,来提高SNP和CNV的阳性率和进一步验证以上推测。
4 结 论
本研究分析了马属动物三成员家系之间结构基因组的遗传多样性,识别了骡异常高的denovoSNP突变率,和骡的特异性CNV。大部分突变相关基因与机体的免疫过程相关。结果说明,马和驴杂交作为一种应激,可能触发骡基因组发生高频率的突变。另外,这些突变可能反映了马和驴的遗传不相容位点,也可能对骡的适应性具有重要意义。这些结构变异的识别,为进一步开展马和驴异种杂交的遗传基础及其分子机制的研究提供候选遗传位点。
