基于全新药物设计方法的木姜子属酰胺类化合物5-LOX抑制剂筛选研究
2023-11-08夏侯真如薛孟祺汪欣怡字慧敏文政琦李碧桃杨靖华
夏侯真如, 薛孟祺,汪欣怡,字慧敏,文政琦,李碧桃*,杨靖华*
基于全新药物设计方法的木姜子属酰胺类化合物5-LOX抑制剂筛选研究
夏侯真如1, 薛孟祺1,汪欣怡2,字慧敏1,文政琦3,李碧桃3*,杨靖华1*
1. 云南大学教育部自然资源药物化学重点实验室,云南省天然产物转化与应用重点实验室,云南大学化学科学与工程学院,云南 昆明 650091 2. 昆明医科大学第一临床医学院,云南 昆明 650500 3. 昆明医科大学第一附属医院,云南 昆明 650500
采用基于分子对接及从头设计的方法从木姜子属酰胺类化合物中寻找潜在的5-脂氧合酶(5-lipoxygenase,5-LOX)抑制剂并改造提高其预期活性。通过文献资料收集了木姜子属植物中现已分离鉴定的酰胺类化合物10个。应用分子对接技术,通过与5-LOX抑制剂筛选相关的蛋白结晶的对接分析,筛选出木姜子属植物中最优的酰胺类化合物。再以该化合物为基础进行分子库扩充,并通过对该分子库进行类药规则和ADMET筛选,选择其中排名前五的分子进行从头设计碎片生长,最后再从设计结果中筛选出更优的5-LOX潜在抑制剂。通过分子对接分析筛选出了木姜子属植物中最优的酰胺化合物-反式-3,4-亚甲基肉桂酰-3-甲氧基酪胺,利用类药规则和ADMET筛选出9个相对符合药理毒理性质要求的类似分子,采用从头设计方法筛选了出打分值较高的候选化合物6个,并初步阐释了优良5-LOX抑制剂可能的活性残基结合情况。木姜子属中酰胺类化合物及其衍生物可能通过抑制5-LOX来发挥抗炎活性作用,相对于传统筛选和改造,分子对接和从头设计技术节约了大量的资源,为新型5-LOX抑制剂的研发提供了一定的参考。
木姜子属;酰胺类化合物; 5-LOX抑制剂;分子对接;从头设计;-反式-3,4-亚甲基肉桂酰-3-甲氧基酪胺
炎症是机体必不可少的一种免疫反应,是机体应对外界刺激的一种自动防御机制。一般来说,炎症的发生对机体是有益,它能够在各种不利条件下维持组织内的稳态。但另一方面,其是以短暂的组织功能下降为代价,这又可能导致体内平衡的改变。因此炎症是机体与致炎因子之间损伤与抗损伤的斗争,炎症的病变以抗损伤反应为主,但也会对机体造成不同程度的损伤[1]。
研究表明,许多炎症反应过程实质上主要是由于一系列被称作炎症介质的内源性化学因子或生物大分子介导来实现的,它们影响到整个炎症过程。所谓炎症介质是指在致炎因子作用下,由局部组织细胞释放,或来源于血浆,参与并诱导炎症发生发展的生物活性物质[2]。白细胞三烯(leucotriene,LT),即白三烯(LTA4、LTB4、LTC4、LTD4、LTE4、LTF4等)是一种非常重要的炎症介质,其中LTC4、LTD4、LTE4因均含有1个半胱氨酸残基(cysteine),故统称为cysLTs,被认为是过敏反应的慢反应物,在炎症级联反应中具有重要作用地位。脂氧合酶(lipoxygenase,LOX)是白三烯合成中最为重要的酶,其在人体中包含5-LOX、15-LOX等亚型,其中15-LOX主要是通过最终催化形成脂氧素来调节炎症反应,而5-LOX则是炎症反应中白三烯形成的主要来源[3]。目前5-LOX作为炎症反应中的重要靶点,已有针对该靶点的药物上市。其中已上市的药物如齐留通和ABT-761等在临床上对炎症反应,尤其是哮喘类疾病已显示出良好的治疗效果,但其有一定的肝毒性[4],且半衰期很短,药动学性质也较为不利[5]。因此,新型5-LOX抑制剂的研究越来越受到人们的关注。
木姜子属Lam.植物作为樟科(Lauraceae)常绿乔木或灌木中种类最多的属之一,在我国南方和西南温暖地区有广泛的分布[6]。木姜子属植物在民间有较为悠久的用药历史,在传统医学中被用于治疗风寒湿痛、跌打损伤、糖尿病等各种疾病,其中抗炎疗效在医书中具有广泛的记载,如山鸡椒作为荜澄茄使用,具有温中散寒、消肿止痛的功效;豺皮樟具有对关节炎、腰腿疼痛的良好疗效;木姜子则可用于治疗风寒湿痛、跌打止痛等[7]。针对木姜子属植物的化学成分,国内外学者已经展开过一些研究[8]。研究表明,木姜子属植物中的酰胺类具有良好的抗炎活性,比如Lee等[9]的动物实验验证了15个酚酰胺能够通过抑制脂质过氧化反应,增强抗氧化酶活性,来降低肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)和核因子(nuclear factor-kB,NF-kB)从而降低一氧化氮的生成。由于在前期分子对接筛选研究中发现木姜子属植物中的酰胺类对5-LOX靶点具有良好的抑制活性,因此本研究选用与5-LOX抑制剂筛选相关的3种不同来源的蛋白结晶,对木姜子属植物中的酰胺类化学成分进行分子对接分析,筛选出其中最优的酰胺类化合物,并以其为骨架进行分子库扩充。再通过对该分子库进行类药规则和ADMET筛选,并以筛选结果前5位的分子进行从头设计的碎片生长,最后从中筛选出更优的5-LOX潜在抑制剂,并初步阐释其活性残基结合情况。
1 材料与方法
1.1 平台与软件
本研究所有工作均在Microsoft Windows 10 Professional操作系统中完成,采用ChemOffice Professional 20.0程序(珀金埃尔默公司)中的ChemBioDraw模块和ChemBio 3D模块、Molegro公司的Molegro Virtual Docker 6.0(MVD)软件、Accelrys公司的Discovery Studio 2016(DS)。参数设置除特殊指明外均为默认值。
1.2 分子对接
1.2.1 受体数据库的组建 从RCSB PDB数据库(http://www.rcsb.org/)搜索并下载与5-LOX抑制剂筛选相关的3种不同来源的蛋白结晶,分别针对大豆sLOX-3(1JNQ)、哺乳动物15-LOX(1LOX)、智人源5-LOX(3V99)[10],其结构PDB编号及原配体与主要靶点残基作用情况见表1。这3种蛋白结晶获取相对容易,有助于与后期活性验证进行匹配,其中大豆同工酶(sLOX-1、sLOX-2、sLOX-3)是药物筛选常见酶,研究发现sLOX-1与智人源LOX酶有较高的氨基酸序列同一性,其有些抑制剂在抑制智人源LOX酶上更为有效[11],但其PDB数据库中晶体结构(1YGE)缺少原配体复合,因此选择相似的sLOX-3晶体结构(1JNQ)作为研究对象,并且Somvanshi等[12]报道了姜黄素等已知抑制剂与sLOX-3结合的解理常数和与智人源5-LOX的较为相似,有助于增加确定抑制剂的可能性;哺乳动物15-LOX的氨基酸序列中与智人源5-LOX具有类似的疏水腔,这在所有的花生四烯酸催化酶中都是保守的,而与5-LOX的特异性区域相比较,有助于确定抑制剂的选择性;智人源5-LOX的PDB数据库相关晶体结构较多,这里选用具有配体复合物的3V99,其与稳定纯酶结晶(3O8Y)具有足够的结构相似性,抑制剂留有足够的空间停靠在活性中心腔中[10]。对这3种蛋白质晶体结构进行删除水分子和辅因子的处理,经Molegro Virtual Docker 6.0(MVD)软件进行氨基酸残基修补,并选择对应原配体位置为中心,选择半径1.2 nm的残基为活性残基,并将其定义为结合口袋空腔,最后保存为PDB格式备用。
1.2.2 配体数据库的组建 通过文献检索收集,已知目前从木姜子属植物中分离得到的酰胺类化合物共有10个[8](图1),将其二维结构绘入ChemBioDraw软件中,并通过ChemBio 3D将其转化为三维结构,并采用MMFF94算法对其能量进行最低优化,其中最大迭代次数设置为5000次,最大RMS梯度设置为0.5,保存为mol2格式作为分子对接的配体分子集。

表1 原配体与主要靶点残基作用情况

图1 木姜子属植物中分离获得的酰胺类化合物
1.2.3 对接方法可行性验证 Molegro Virtual Docker 6.0(MVD)在半柔性对接精度方面高于市面上的绝大多数软件,在于其能够通过不同打分函数和搜索算法的组合来获取针对受体蛋白的对接方法[13]。因此,将含有原配体的蛋白晶体复合结构中的原配体抽离,然后利用ChemBio 3D中的MMFF94算法对其进行能量最低优化,再按照设定参数对接回原结合口袋,基于不同打分函数和搜索算法的组合,反复进行尝试,从而获取重现性较好的对接方法。
1.2.4 分子对接参数设置 将上述组建完成的受体和配体分别导入Molegro Virtual Docker 6.0(MVD)中,并设置配体始终保持柔性,搜索算法运行次数100次,其余参数保持不变。靶点蛋白1JNQ结合空腔坐标位置为X:20.36、Y:3.30、Z:19.87;靶点蛋白3V99结合空腔坐标位置为X:18.38、Y:−78.70、Z:−33.89;靶点蛋白1LOX结合空腔坐标位置为:X:−27.12、Y:151.87、Z:56.97。
1.2.5 结果处理 对接完成后,分别以靶点蛋白中原有配体为参照,对接结果依据Rerank Score(R-Score)评价。在对接时,为了保证打分结果的可靠性,设置得到每个分子对接结果的前3位,并取其平均值作为最终得分。R-Score数值为负值,表征分子与蛋白能够结合,数值越小,表征亲和性越大[14-15]。根据R-Score结果,筛选出每个靶点蛋白得分排名靠前的化合物。通过对比分析,获取对3种来源的靶点蛋白均有很好的打分数值的化合物。再以此化合物为基本骨架,通过Zinc 15数据库(http://zinc.docking.org/)进行分子库扩充,获取类似化合物分子库。
1.3 类药规则及ADMET筛选
将以分子对接获取的化合物为基本骨架得到的类似化合物分子库,转化为3D结构导入Discovery Studio 2016(DS),调用Filter by Lipinski and Veber Rules模块,勾选Lipinski Rule of Five 和Veber Rule两种规则对该分子库进行筛选。再根据筛选结果,调用ADMET descriptors模块,勾选aqueous solubility、blood brain barrier penetration、CYP2D6 binding、hepatotoxicity、intestinal absorption、plasma protein binding等6个筛选方向,最终获取相对符合药理毒理性质要求的化合物[16-17]。
1.4 从头设计
1.4.1 对接准备 为了减少软件间的系统性误差,因此从头设计的对接筛选工作将由DS 2016进行实现。以通过类药规则和ADMET筛选的化合物作为配体,智人源(Homo sapiens)5-LOX靶点蛋白3V99作为受体,导入DS 2016,并用Prepare Ligands和Clean Protein模块分别对配体和受体进行处理,然后用Define Receptor模块将蛋白分子3V99定义为受体蛋白,并利用Form Current Selection模块设置原配体所在位置为活性空腔中心,半径为1.2 nm,最后调用CDOCKER模块对接模块,设置Pose Cluster Radius为0.2 nm,以保证与MVD软件对接参数的一致性,且使对接构象尽可能具有多样性,其余参数保持默认[18],最终对接结果仅保留非离子型构象。
1.4.2 碎片生长 对接完成后,以原有配体为参考,对接结果依据-CDOCKER ENERGE进行评价,-CDOCKER ENERGE数值越大则对接结果越好,以每个配体打分最高的构象的数值作为依据。选取对接结果排名前5的化合物分别进行分子碎片生长,调用De Novo Evolution模块,选取默认碎片库进行碎片生长,共获取50个新化合物,每组10个[19-20]。
1.4.3 结果筛选 将碎片生长所得的50个新化合物作为配体,智人源(Homo sapiens)5-LOX靶点蛋白3V99作为受体,在DS 2016中使用CDOCKER模块进行分子对接筛选,Pose Cluster Radius设置为0.2 nm。对接完成后,同样以原有配体为参考,对接结果依据-CDOCKER ENERGE进行评价。
2 结果
2.1 分子对接
2.1.1 对接方法可行性验证结果选取的3个靶点均为含有原配体的蛋白晶体结构,分别将原配体抽离,再对接回原有的结合口袋,计算对接后构象与原配体结构的均方根偏差值(RMSD值),数值小于0.2 nm,说明该对接方法针对于此受体蛋白和参数设定可用[21]。为了减少系统性误差,本次均使用打分函数MolDock Score[GRID]和搜索算法GPU Screening(CUDA)的组合对接方法。
2.1.2 对接结果木姜子属植物中分离得到的酰胺类化合物针对与5-LOX抑制剂筛选相关的3种不同来源的蛋白结晶1JNQ、1LOX、3V99的筛选结果见表2。通过对比3种不同来源蛋白结晶的筛选结果可发现化合物6的打分结果均在前3位以上,且优于原配体和上市药物齐留通。以化合物6为例,展示其与3种蛋白结晶的2D相互作用图,并通过与原配体和齐留通的2D作用图(图2)进行对比,结果表明主要的结合残基和作用力类似,可以发现3者的结合模式较为相似。

表2 对接得分结果
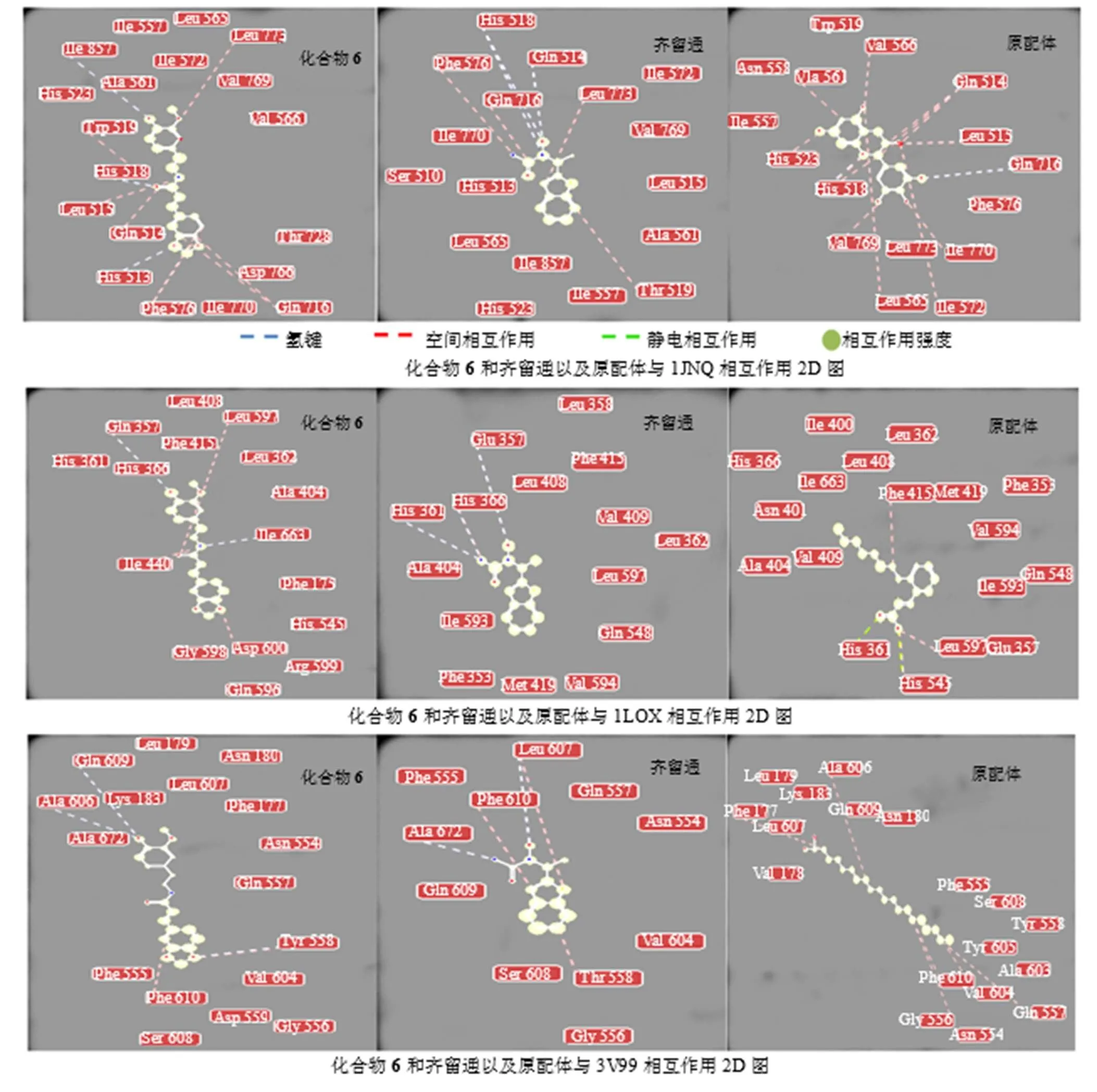
图2 化合物6和齐留通以及原配体与3种蛋白受体相互作用2D图
2.2 类药规则及ADMET筛选
以化合物6为基本骨架得到的231个编号为Z001~Z231的类似化合物组成的分子库,导入DS 2016进行类药规则和ADMET筛选。化合物全数符合药理性质要求,后经过ADMET筛选,筛选表格依据见表3,仅展示相对最优的筛选结果,整体结果ADMET plot见图3,仅显示血脑屏障通透性(blood brain barrier penetration,BBB)模型和人体肠道吸收性(human intestinal absorption,HIA)模型95%和99%的置信区间,图中每个点代表1个预测分子,位于99%置信区间以外的分子被认为是不可靠的,最终得到9个相对符合毒理性质要求的化合物,筛选结果见图4。

表3 ADMET筛选结果*
*人体肠道吸收性:0~3吸收性依次递减;水溶性(25 ℃):0~6溶解性依次增大,4为最好情况;血脑屏障通透性:0~5通透性依次降低; TURE or FALSE-是或否
*human intestinal absorption: the absorptivity decreased from 0 to 3; water solubility (25℃): the solubility increases from 0 to 6, with 4 being the best case; Blood-brain barrier permeability: permeability decreased from 0 to 5; TURE or FALSE- yes or no
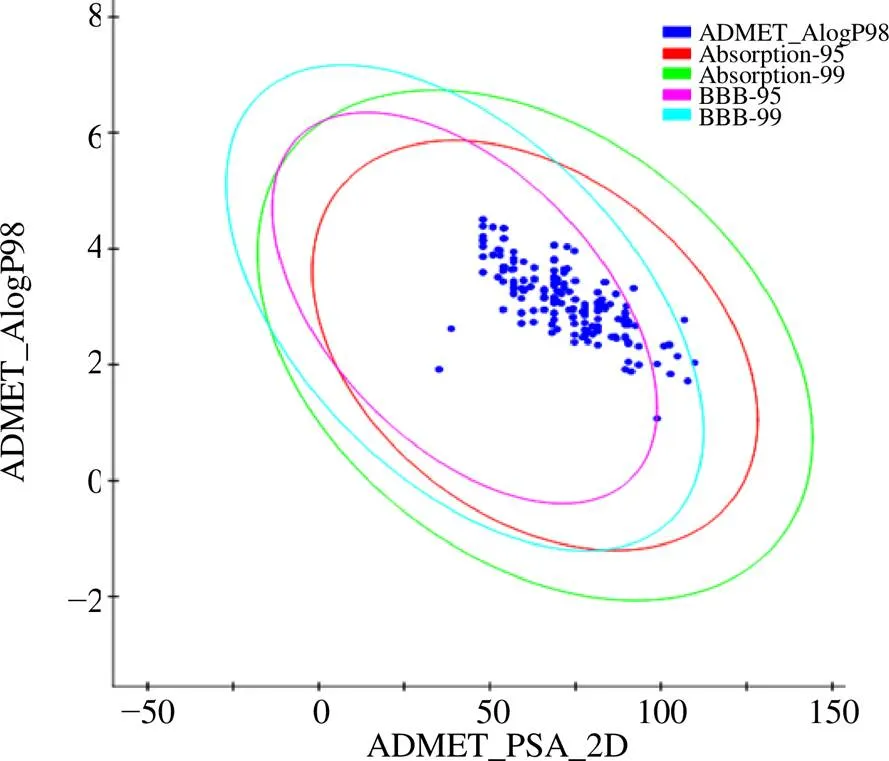
图3 ADMET筛选图
2.3 从头设计
在 DS 2016中,将相对符合药理毒理性质要求的9个化合物作为配体,智人源(Homo sapiens)5-LOX靶点蛋白3V99作为受体,进行CDOCKER模块的分子对接筛选,见表4,并选取-CDOCKER ENERGE数值排名前5编号为Z167、Z184、Z180、Z125、Z004的化合物作为分子骨架,调用De Novo Evolution模块进行碎片生长,获取5组化合物共50个。再以这50个化合物为分子库,进行分子对接筛选,获取潜在的活性分子,见表5。从表5中可以看出-CDOCKER ENERGE数值最高的为Z167-6,考虑实际价值,筛选结果仅展示排名前6的分子(除离子型),其中数值高于原配体的分子共有1个,而高于齐留通的分子共有2个(图5)。考虑到排名较高的分子均来自与Z167和Z180的碎片生长,因此展示2组分子中排名前3的分子Z167-6、Z167-4、Z180-5及排名靠后(负值除外)的分子Z167-7、Z180-3、Z180-1与受体蛋白3V99的相互作用2D图,并与齐留通和原配体的2D作用图进行分析比较见图6。
3 分析与结论
3.1 分子对接
从3种不同来源蛋白结晶的分子对接筛选结果中发现,木姜子属中酰胺类化合物6的Rerank Score打分结果均在前3位以上,且优于原配体和上市药物齐留通的打分值。并且从2D相互作用图中可以发现,在1JNQ中,化合物6与蛋白中的活性残基His518可以形成氢键,与Gln716、Leu716、Leu773等可以形成较强的空间相互作用,其结合模式与齐留通和原配体均类似;在1LOX中,活性残基Leu362、Leu367、Ile350、Leu597和Ile400组成的疏水腔在所有花生四烯酸催化酶和其类似物中是保守的[10],而化合物6与蛋白中的活性残基Glu357可以形成氢键,与Leu597、Ile400具有强空间相互作用,Leu362具有较弱空间相互作用,这相较于齐留通和原配体与相应残基的作用情况要强;在3V99中,活性残基Tyr181、Ala603、Ala606、His600、Thr364和Phe177被认为是5-LOX的特异性区域,化合物6与蛋白中的活性残基Ala606、Gln609可以形成氢键,与Phe177具有空间相互作用,相较于齐留通和原配体的作用情况要强。木姜子属植物中酰胺化合物6具有与齐留通和原配体相似的活性残基作用模式,除此之外其与文献报道的分子对接相关活性残基作用方式类似[10],并且对于3种蛋白的对接打分数值皆排名靠前。因此推测其可能表现有相关的LOX酶抑制活性作用,特别是对智人源5-LOX有较大的抑制可能性,因此以化合物6为基础扩展分子库获取了具有相似结构的231个化合物,其中9个化合物通过了类药规则和ADMET规则筛选,相对符合药理毒理性质要求。
图4 类药规则及ADMET筛选结果

表4 9个化合物分子对接筛选结果
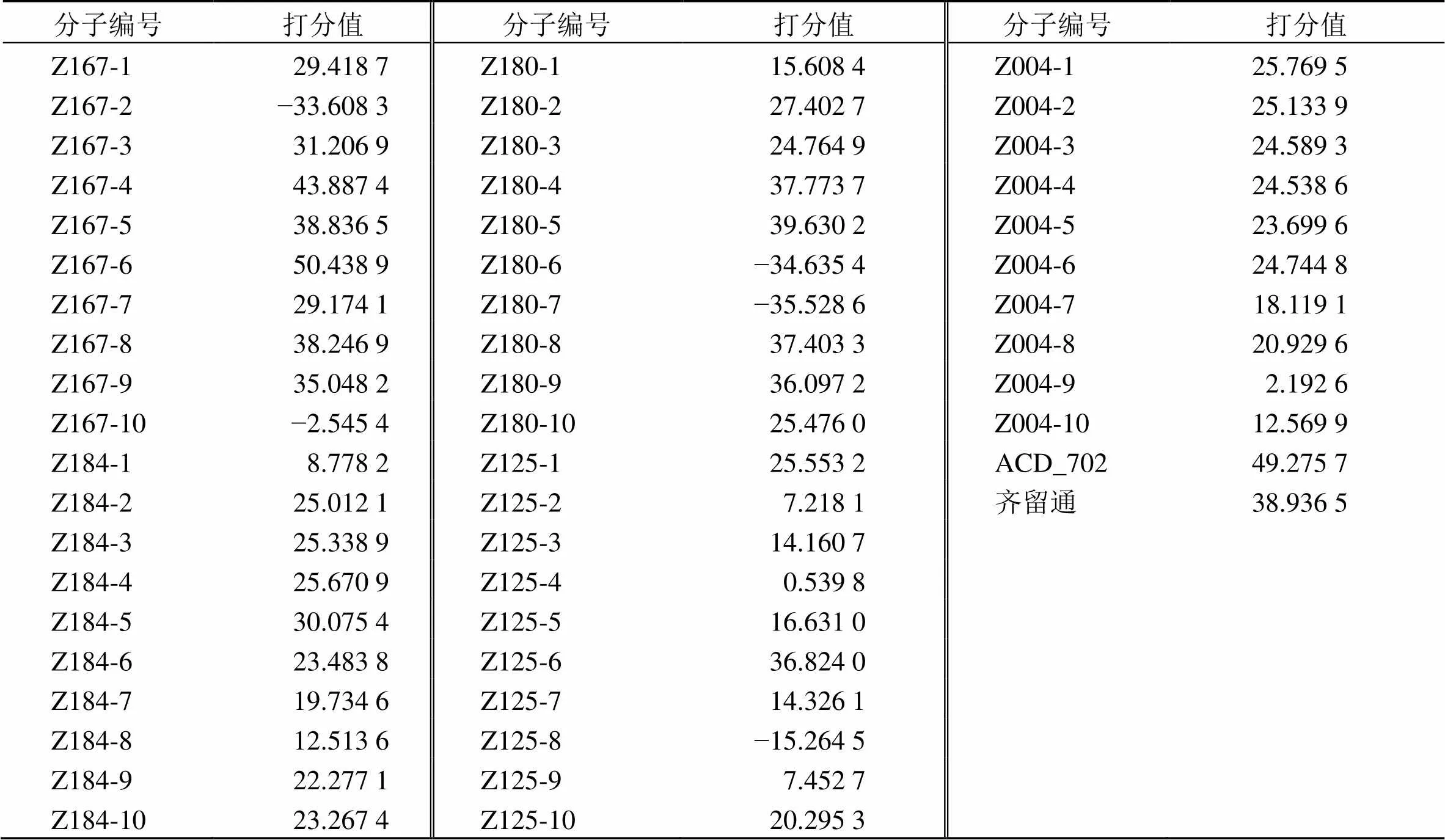
表5 从头设计筛选结果

图5 打分值排名前6的分子(除离子型)

图6 3V99相互作用2D图
3.2 从头设计
在对5-LOX的从头设计研究中,选取了上述9个相对符合药理毒理性质要求的化合物中-CDOCKER ENERGE数值排名前5的化合物Z167、Z184、Z180、Z125、Z004作为分子骨架,其打分数值均在阳性对照齐留通附近,其中Z167、Z184的打分数值高于齐留通。经过从头设计碎片生长后,Z167分子衍生物整体打分数值有较大幅度的提升,其余分子衍生物整体打分数值多有下降。因此对整体分数较高的2组衍生物分子2D相互作用情况进行分析,选择其中排名前3的分子和排名靠后的分子(负值除外)展示。结果显示打分数值较优的衍生物分子或与蛋白结晶3V99的活性残基Ala672有Pi-Anion的静电相互作用,或与His372有Carbon Hydrogen Bond的氢键作用、或与His367有Pi-Alkyl疏水作用,这与齐留通和原配体的作用模式类似,而Ala410、Lys409等形成的疏水作用、Gln363形成的氢键作用可能起到稳定作用,从而提高了打分数值。打分数值较低的衍生物分子也部分显示了以上作用情况,进一步证实了优良5-LOX抑制剂的结合位点活性残基可能与上述有关。综上所述,可以推测优良5-LOX抑制剂的开发关键点在于与上述活性残基的结合,其真实药理药效作用及活性情况正待验证中。
现代药物研究中,分子对接方法和从头设计方法作为基于受体的药物设计方法是计算机辅助药物设计中应用广泛且精度较高的方法。它们通过化学计量学方法模拟药物分子与蛋白受体之间的作用关系,快速大量筛选得到潜在药物分子,并根据结合口袋情况针对性进行分子碎片生长,从而得到更为精准结合力更高的潜在药物分子[22-23]。本研究通过分子对接分析筛选出木姜子属植物中酰胺类化合物里最可能拥有5-LOX潜在抑制活性的分子,通过类药规则和ADMET规则筛选出9个类似骨架的相对符合药理毒理性质要求的分子,最终通过从头设计碎片生长预测了结合力更高更具活性潜力的衍生物分子,并推测了5-LOX抑制剂的关键结合活性残基情况,从而为研发5-LOX抑制剂类药物提供了一定的参考。
利益冲突 所有作者均声明不存在利益冲突
[1]Medzhitov R. Inflammation 2010: New adventures of an old flame [J]., 2010, 140(6): 771-776.
[2]Ogawa Y, Calhoun W J. The role of leukotrienes in airway inflammation [J]., 2006, 118(4): 789-798.
[3]Sharma J N, Mohammed L A. The role of leukotrienes in the pathophysiology of inflammatory disorders: Is there a case for revisiting leukotrienes as therapeutic targets? [J]., 2006, 14(1): 10-16.
[4]Joshi E M, Heasley B H, MacDonald T L. 2-ABT-S-oxide detoxification by glutathione S-transferases A1-1, M1-1 and P1-1: Implications for toxicity associated with zileuton [J]., 2009, 39(3): 197-204.
[5]Braeckman R A, Locke C S,. The pharmacokinetics of zileuton in healthy young and elderly volunteers [J]., 1995, 29(2): 42-48.
[6]李锡文. 中国植物志(第31卷) [M]. 北京: 科学出版社, 1982: 261.
[7]谢宗万, 余友芩. 全国中草药名鉴 [M]. 北京: 人民卫生出版社, 1996: 114-117.
[8]Wang Y S, Wen Z Q, Li B T,. Ethnobotany, phytochemistry, and pharmacology of the genus: An update [J]., 2016, 181: 66-107.
[9]Lee Y T, Hsieh Y L, Yeh Y H,. Synthesis of phenolic amides and evaluation of their antioxidant and anti-inflammatory activityand[J]., 2015, 5(104): 85806-85815.
[10]Tsolaki E, Eleftheriou P, Kartsev V,. Application of docking analysis in the prediction and biological evaluation of the lipoxygenase inhibitory action of thiazolyl derivatives of mycophenolic acid [J]., 2018, 23(7): 1621.
[11]Lapenna D, Ciofani G, Pierdomenico S D,. Inhibitory activity of salicylic acid on lipoxygenase-dependent lipid peroxidation [J]., 2009, 1790(1): 25-30.
[12]Somvanshi R K, Singh A K, Saxena M,. Development of novel peptide inhibitor of Lipoxygenase based on biochemical and BIAcore evidences [J]., 2008, 1784(11): 1812-1817.
[13]马宏跃, 张启春, 周婧, 等. 黄芩中四种主要黄酮成分与环氧酶-2的分子对接研究 [J]. 中医药学报, 2009, 37(6): 71-74.
[14]Xu S K, Peng H, Wang N,. Inhibition of TNF-α and IL-1 by compounds from selected plants for rheumatoid arthritis therapy:andstudies [J]., 2018, 17(2): 277.
[15]Santosa H, Putra G S, Yuniarta T A,. Synthesis and molecular docking studies of’-benzoylsalicylhydrazide derivatives as antituberculosis through InHA enzym inhibition [J]., 2018, 29(4): 198.
[16]van de Waterbeemd H, Gifford E. ADMET in silico modelling: Towards prediction paradise? [J]., 2003, 2(3): 192-204.
[17]Ajay D, Sobhia M E. Identification of novel, less toxic PTP-LAR inhibitors using in silico strategies: Pharmacophore modeling, SADMET-based virtual screening and docking [J]., 2012, 18(1): 187-201.
[18]谷宇, 张栩, 陈艳昆, 等. 基于分子模拟技术筛选大黄、羌活、秦艽的5-LOX、LTA4H抑制剂 [J]. 中国中药杂志, 2017, 42(23): 4494-4502.
[19]吴景卫. 4-噻唑烷酮类CDC25抑制剂的设计与合成 [D]. 天津: 天津医科大学, 2015.
[20]Jiang Y Y, Gao H W. Pharmacophore-based drug design for potential AChE inhibitors from traditional Chinese medicine database [J]., 2018, 76: 400-414.
[21]Rao S N, Head M S, Kulkarni A,. Validation studies of the site-directed docking program LibDock [J]., 2007, 47(6): 2159-2171.
[22]谢治深, 宋军营, 张振强, 等. 计算机辅助药物设计方法及其在新药研发中的应用 [J]. 河南大学学报: 医学版, 2019, 38(2): 148-152.
[23]翟红梅, 曹长春, 韩永红, 等. 从头药物设计技术 [J]. 北方药学, 2013, 10(2): 69.
Vitual screening of 5-LOX inhibitors of amide compounds from Litsea plants based on novel drug design method
XIA HOU Zhen-ru1, XUE Meng-qi1, WANG Xin-yi2, ZI Hui-min1, WEN Zheng-qi3, LI Bi-tao3, YANG Jing-hua1
1. Key Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education; Yunnan Provincial Key Laboratory of Natural Product Transformation and Application; School of Chemical Science and Technology, Yunnan University, Kunming 650091, China 2. No. 1 School of Clinical Medicine, Kunming Medical University, Kunming 650500, China 3. First Affiliated Hospital of Kunming Medical University, Kunming 650500, China
To find potential 5-lipoxygenase (5-LOX) inhibitors and improve the expected activity of amide compounds fromplants based on molecular docking and de novo design.A total of ten amide compounds isolated and identified fromplants were collected in the literature. The molecular docking technique was used to screen the optimal amide compound inplants by docking analysis of protein crystals associated with 5-LOX inhibitor screening. Then expanding the molecular library based on the compound, selecting the top five molecules to perform de novo design fragment growth by performing drug-like rule and ADMET screening on the molecular library, and finally screening out a more optimal 5-LOX potential inhibitor from the design result.The optimal amide compound--3,4-methylenecinnamyl-3-methoxytyramine inplants was screened out by molecular docking analysis. A total of nine similar molecules relatively meeting the requirements for pharmacological and toxicological properties were screened out by the drug-like rule and ADMET. A total of six candidate compounds with high scoring values were screened out by the de novo design method, and the possible active residue binding of excellent 5-LOX inhibitors was preliminarily explained.Amides and their derivatives in theplants may exert anti-inflammatory activity by inhibiting 5-LOX. Compared with traditional screening and modification, molecular docking and de novo design technology save a lot of resources. This study provides a certain reference for the research and development of new 5-LOX inhibitors.
Lam.; amide compounds; 5-LOX inhibitor; molecular docking; de novo design;--3,4-methylenecinnamyl-3-methoxytyramine
R284
A
0253 - 2670(2023)21 - 6961 - 10
10.7501/j.issn.0253-2670.2023.21.005
2023-03-09
国家自然科学基金资助项目(82160661);国家自然科学基金资助项目(81960629);国家自然科学基金资助项目(21662040);云南省创新团队(202005AE160005);云南省应用基础研究项目(2017FD122)
夏侯真如(1995—),男,安徽人,在读硕士,研究方向为天然药物化学。Tel: 18325790633 E-mail: 546552125@qq.com
通信作者:李碧桃(1971—),女,云南人,主任医师,主要从事儿童体格生长发育、儿童营养、儿童神经心理发育、儿童早期发展研究工作。E-mail: 1827023387@qq.com
杨靖华(1971—),女,云南人,博士,教授,主要从事天然药物化学研究工作。E-mail: yangjh@ynu.edu.cn
[责任编辑 王文倩]
