杏仁核N-甲基-D-天冬氨酸受体在芬太尼诱发痛觉过敏大鼠中的作用
2023-11-07尹平平许睿周张胡晓榆于明良
尹平平 许睿 周张 胡晓榆 于明良
(武汉市第四医院麻醉科,湖北 武汉 430034)
芬太尼是人工合成的阿片受体激动剂,与其他阿片类药物一样,具有强效的镇痛作用,但重复使用会引起机体对疼痛的敏感性增强,即阿片诱发的痛觉过敏(OIH)〔1〕,这种副作用严重干扰了临床医生对疼痛的管理和治疗〔2〕,亟待探明其发生机制。N-甲基-D-天冬氨酸受体(NMDAR)广泛存在于中枢神经系统,包括杏仁核(CeA),且NMDAR与疼痛的发生密切相关〔3〕。研究表明,脊髓NMDAR可被细胞外调节蛋白激酶(ERK)激活,使NMDAR磷酸化增加,导致其功能增强,最终参与炎性OIH的产生〔4〕,而下调脊髓NMDAR的表达可以减弱瑞芬太尼诱导的OIH〔5〕。研究证明,CeA内ERK与OIH的产生有关〔6,7〕,但CeA内NMDAR在OIH中的作用尚待进一步研究。因此,本研究拟探讨CeA内NMDAR在芬太尼诱发的OIH大鼠中的作用,以便为临床预防和治疗OIH提供思路。
1 材料与方法
1.1动物选择 健康清洁级(SPF级)雄性SD大鼠36只,体质量70~110 g,由华中科技大学同济医学院附属同济医院动物实验中心提供,室温饲养,自由摄食和饮水,白天与黑夜时间12 h∶12 h。本实验所用的大鼠均严格遵循中国卫生机构制定的实验室动物应用指南。
1.2实验分组 实验一:采用随机数字表法,将24只SD大鼠随机分为3组(n=8):芬太尼+氯胺酮组(K1组)皮下注射芬太尼制备OIH模型,1 d后腹腔注射NMDAR拮抗剂氯胺酮(15 mg/kg);芬太尼诱发OIH组(H1组)皮下注射芬太尼制备OIH模型,1 d后腹腔注射等容量生理盐水;对照组(C1组)皮下注射等容量生理盐水,1 d后腹腔注射等容量生理盐水。分别于皮下注射芬太尼或生理盐水前(T0)、注射后1 d(T1)和腹腔注射氯胺酮或生理盐水后30 min(T2)时检测大鼠机械痛阈和热痛阈。实验二:将实验一最后一次测痛结束的各组大鼠处死,取右侧CeA区组织,Western印迹检测NMDAR亚基NR2B磷酸化(P-NR2B)的表达。实验三:另取12只雄性SD大鼠,随机分为3组(n=4):对照组(C2组)、芬太尼诱发OIH组(H2组)和芬太尼+氯胺酮组(K2组)按实验一处理后,制备含有中央CeA的脑片,采用CeA区神经元电生理记录各组右侧CeA区神经元NMDAR微小兴奋性突触后电流(mEPSCs)的幅值和频率。
1.3芬太尼诱发OIH模型的制备 枸橼酸芬太尼(湖北宜昌人福药业有限公司,批号1130411)使用前用生理盐水配制。采用文献〔8〕的方法制备芬太尼诱导的OIH模型。具体操作是:大鼠颈背部皮下注射芬太尼,每次60 μg/kg,共4次,给药间隔15 min,累积药量240 μg/kg。给药期间及给药后2 h内注意给大鼠保暖及吸氧。
1.4疼痛行为测定 分别于T0、T1和T2时测定大鼠的机械痛阈和热痛阈。测定机械痛阈时,具体方法是将大鼠置于金属笼内,静置30 min后,用不同压力的von Frey丝(North Coast公司,美国)垂直刺激大鼠左足掌面,力度以von Frey丝轻微弯曲为准,持续10 s或直至出现缩足反应。当大鼠在规定时间内出现缩足反应,则记为阳性。采用up-and-down方法计算50%缩足反应阈即为机械痛阈值。测定热痛阈时,采用的热痛刺激仪(BME-410C)购于中国医学科学生物医学工程研究所。具体方法是将大鼠置于底部为玻璃板的透明小室中,安静放置30 min后,使用辐射灯对准大鼠左足掌正中照射,自动记录大鼠的缩足潜伏期作为热痛阈,停止时间为15 s,以免造成大鼠左足掌组织损伤。
1.5Western印迹检测CeA组织P-NR2B的表达 测痛完毕后处死大鼠,并取出右侧CeA区组织冻存于-80 ℃冰箱备用。取适量组织称重后,加入一定量含有蛋白酶抑制剂的RIPA裂解液(碧云天,中国)并匀浆,12 000 r/min,4 ℃离心10 min取上清,二喹啉甲酸(BCA)法测蛋白浓度。加入十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)上样缓冲液(碧云天,中国),95 ℃煮沸8~10 min使蛋白变性,制备SDS-PAGE(10%),电泳后湿转至聚偏氟乙烯(PVDF)膜上。5%脱脂奶粉TBST缓冲液室温震荡封闭1 h后洗膜,加入封闭液稀释的兔抗P-NR2B抗体 (1∶500稀释,Millipore公司,德国)和小鼠抗GAPDH(1∶500,博士德,中国),4 ℃孵育过夜,洗膜,加入TBST稀释的辣根过氧化酶(HRP)-山羊抗兔和HRP-山羊抗小鼠的二抗(1∶5 000),室温孵育2 h,洗膜后在避光条件下将电化学发光(ECL)试剂盒中的A、B两种试剂等体积混合,振荡混匀后均匀加到膜上,用化学发光凝胶图像系统拍照。P-NR2B表达量以P-NR2B与GAPDH光密度的比值表示。
1.6全细胞膜片钳实验 全细胞膜片钳技术记录大鼠CeA区神经元NMDAR mEPSCs。采用LEICA VT1000 S切片机,将含有CeA区的大鼠右脑组织块冠状切成厚度为350 μm的脑片。切片液成分(mmol/L):213蔗糖,3 KCl,1 NaH2PO4,0.5 CaCl2,5 MgCl2,26 NaHCO3和10葡萄糖,切片液温度为4 ℃。将切片所得的含有CeA区的脑片在25 ℃的人工脑脊液(ACSF)中孵育,时间至少1 h,人工ACSF成分(mmol/L):125 NaCl,5 KCl,1.2 NaH2PO4,2.6 CaCl2,1.3 MgCl2,26 NaHCO3和10葡萄糖。其中切片液及人工ACSF均为氧饱和溶液,且调节PH至7.2~7.4,渗透压维持在290~310 mOsm。使用的持续灌流ACSF的温度控制在31 ℃,灌流速度为2 ml/min。膜片钳放大器为HEKA EPC-10 (Molecular Devices),记录软件为patchmaster (Molecular Devices),应用红外线微分干涉相差显微镜在可视化模式下记录CeA区神经元mEPSCs。确认封接电阻(>2 GΩ)和串联电阻(<20 MΩ)以保证细胞封接质量。记录电极为玻璃微电极(WPI),电极内液成份(mmol/L):100葡萄糖酸钾、30 KCl、5 NaCl、1 MgCl2、10 HEPES、 3 EGTA、2 Na2-ATP,用1 mmol/L NaOH调节PH值为7.4。持续灌流ACSF中加入 50 μmol/L木防已苦毒素(PTX)阻断抑制性突触后电流,1 μmol/L河豚毒素(TTX,阻断动作电位)和20 μmol/L CNQX(AMPA受体和海人藻受体的拮抗剂),且钳制电压+60 mV的条件下进行CeA区神经元细胞的高阻封接,从而获得NMDA受体mEPSCs。
1.7统计学方法 采用SPSS21.0软件进行方差分析、t检验。
2 结 果
2.1芬太尼诱发的OIH可以被NMDAR拮抗剂所逆转 H1组T1、T2时及K1组T1时机械缩足阀值和热缩足潜伏期均明显低于本组T0时(均P<0.05);与C1组比较,H1组T1、T2时、K1组T1时机械缩足阈值和热缩足潜伏期均明显降低(P<0.05);而给予氯胺酮后可逆转OIH,即与H1组比较,K1组T2时机械痛阈和热缩足潜伏期均明显升高(均P<0.05),见表1。

表1 各组不同时间点机械缩足阈值和热缩足潜伏期比较
2.2芬太尼诱发的OIH与大鼠CeA中P-NR2B的过度表达有关 与C1组CeA区P-NR2B水平(0.28±0.01)相比,H1组(0.48±0.05)明显增多(P<0.05),K1组(0.28±0.02)差异无统计学意义(P>0.05)。见图1。
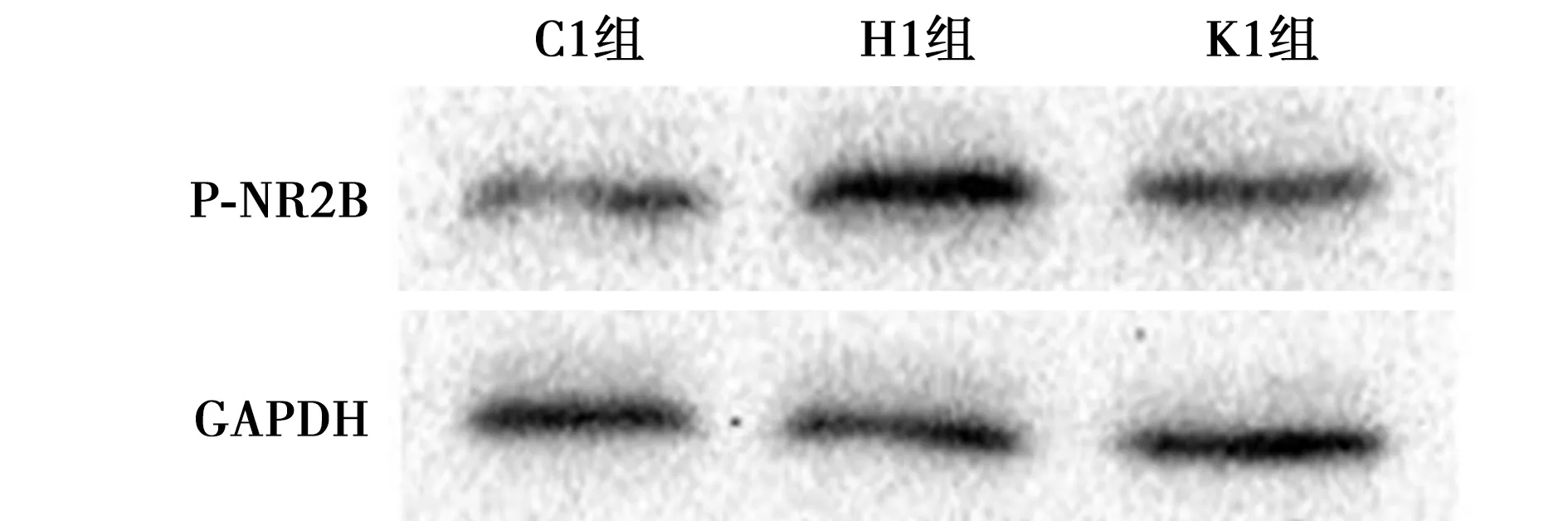
图1 Western印迹检测3组P-NR2B水平
2.3芬太尼诱发的OIH大鼠CeA区神经元NMDAR介导的突触传递发生改变 CeA区神经元电生理记录示,与C2组比较,H2组CeA区NMDAR mEPSCs幅值及频率均明显增加(均P<0.05),而K2组差异均无统计学意义(均P>0.05)。见表2、图2。

图2 各组NMDAR mEPSCs的典型波形(10 s)
表2 各组mEPSCs的幅值和频率比较
3 讨 论
阿片药物芬太尼主要用于临床麻醉和镇痛,但其有潜在的诱发OIH的风险,即采用阿片药物镇痛,却反而引起机体对疼痛的敏感性增加〔9〕。这种矛盾的现象给临床使用阿片类药物带来了困惑,亟待探明其机制,以指导临床用药和治疗OIH。NMDAR广泛存在于中枢神经系统,参与多种疼痛机制的调节〔10〕,包括神经病理性疼痛和中枢痛敏。已有研究表明,脊髓NMDAR参与阿片药物瑞芬太尼诱导的OIH的产生〔11〕,但NMDAR是否参与脊髓上中枢神经系统来调节OIH尚待研究。本研究结果表明CeA NMDAR可能与芬太尼诱发的OIH产生有关。值得注意的是,NMDAR的拮抗剂氯胺酮,除了具有分离麻醉的特性外,还具有镇痛、抗炎和抗抑郁的作用。不同剂量的氯胺酮所产生的作用不同,研究表明,氯胺酮产生麻醉作用所需的剂量远大于其产生镇痛、抗炎和抗抑郁作用所需的剂量〔12〕。鉴于氯胺酮的镇痛作用与给药剂量有关,本研究OIH大鼠单次腹腔注射低剂量(15 mg/kg)氯胺酮,该剂量远低于产生麻醉作用所需的氯胺酮。且氯胺酮小鼠腹腔注射后的半衰期是13~25 min〔13~15〕,在注射30 min后,大鼠并未出现麻醉相关表现,大鼠完全清醒,活动自如,推断氯胺酮此时主要产生的是镇痛作用。在Abelaira等〔16〕研究中,给予大鼠15 mg/kg单次剂量的氯胺酮30 min后,即开始进行大鼠强迫游泳的行为学测试,说明大鼠是完全清醒的,也与本文观察的结果一致。该研究结果提示,NMDAR拮抗剂氯胺酮可以改善OIH大鼠的疼痛阈值,产生镇痛作用,从而缓解OIH。
NR2B是NMDAR执行调节功能的重要亚基,其磷酸化水平与NMDAR介导产生的慢性疼痛密切相关〔17〕,在NR2B亚基激活Ca2+内流之后,特定的鸟嘌呤核苷酸交换因子(GEFs)会刺激ERK的磷酸化(pERK)。pERK随后从神经元细胞质转位到细胞核。在细胞核中,pERK刺激核糖体激酶2,导致133丝氨酸上的环磷腺苷效应元件结合蛋白(CREB)磷酸化(pCREB)。pCREB进一步调节含有CRE的某些基因与CREB结合位点结合。通过与c-fos,神经激肽/P物质受体,脑源性神经营养因子(BDNF),降钙素基因相关肽(CGRP)和环氧化酶(COX)-2等结合,导致伤害性感受反应的产生。这些神经调节剂会引发更多的NR2B亚基的激活,导致中枢神经系统的可塑性改变,这条途径的最终结果是OIH和异常性疼痛的产生。本研究发现,OIH时,NMDAR亚基P-NR2B表达增加,给予NMDAR拮抗剂氯胺酮后,P-NR2B表达降低,可以逆转OIH。
已有研究表明,NR2B亚基在NMDAR调控神经元突触可塑性中起关键性作用,其表达水平与NMDAR产生的兴奋性突触传递正相关〔18〕。本研究中,OIH大鼠CeA NMDAR mEPSCs的幅值和频率增加,给予NMDAR拮抗剂氯胺酮后,可以减少OIH大鼠CeA NMDAR介导的mEPSCs的幅值和频率。mEPSCs的幅值体现的是作用于突触后膜神经递质释放量,而mEPSCs的频率体现的是突触前膜神经递质释放概率,该研究结果提示OIH大鼠CeA神经元间突触传递功能增强,CeA神经元间可能发生了功能上的突触可塑性,且这种改变与NMDAR有关。本研究结果证实了阿片药物芬太尼可能通过某种途径作用于大鼠CeA神经元上的NMDAR亚基NR2B,从而调控神经元间突触传递,使CeA发生突触可塑性,从而产生OIH。但NMDAR是通过何种途径调控大鼠OIH的产生尚待进一步研究。
