PPARα基因敲除诱导小鼠肝脏脂质蓄积的作用研究
2023-11-02秦可欣陈小翠崔元峰阿比旦阿卜杜如苏力托罗娜依米吉提陈邦党
秦可欣, 陈小翠, 崔元峰, 阿比旦·阿卜杜如苏力, 托罗娜依·米吉提, 陈邦党,
(新疆医科大学1基础医学院, 2第一附属医院临床医学研究院, 乌鲁木齐 830000)
非酒精性脂肪肝(Nonalcoholic fatty liver disease,NAFLD)是以肝实质细胞脂肪变性和过量脂质蓄积为主要临床病理学特征的慢性肝病,累及全球25%的人口[1]。1999-2018年间我国NAFLD发病率由18%增加到29%,呈逐年上升且年轻化趋势[2]。NAFLD患者肝脏脂肪酸氧化功能障碍,导致过度异位脂质沉积和细胞内氧化还原平衡改变[3],并产生胰岛素抵抗、炎症和氧化应激反应,促使NAFLD进展为脂肪性肝炎(NASH)、肝纤维化、肝硬化甚至肝细胞癌[4],已逐渐成为肝衰竭和肝移植的重要原因。
过氧化物酶体增殖物激活受体(Peroxisome proliferator-activated receptors,PPARs)是核受体超家族的成员,有三种异构型(α、β/δ和γ亚型)。PPARα是一种配体激活的转录因子,除肝脏外,其在心脏、骨骼肌、棕色脂肪、肠道和肾脏等多种组织中均有表达,参与诱导脂质代谢途径相关基因表达,下调炎症反应有关信号转导途径相关基因表达[5]。临床研究发现肝脏中PPARα基因表达与NASH严重程度、内脏脂肪和胰岛素抵抗呈负相关,且NASH组织学的改善与其表达增加相关[6]。在肝脏特异性PPARα敲除小鼠中,高脂饮食诱导12周其肝脏炎症相关指标和血浆总胆固醇水平明显增高[7]。然而,敲除肠道中PPARα可限制机体脂质的吸收,并限制脂滴的扩张,抑制肝脏组织中的脂质沉积[8]。以上结果体现了PPARα在肝脏脂质代谢稳态和NAFLD进展中的重要地位,但其作用机制较复杂,为了进一步阐明PPARα在机体脂质代谢中的作用,本研究利用全身性PPARα基因敲除小鼠模型,研究PPARα对肝脏脂质蓄积的调节功能,并通过检测肝组织中PPARα所调控的脂肪酸转位酶(CD36)、花生四烯酸ω羟化酶(Cyp4a14)以及脂滴包被蛋白2(Plin2)的蛋白表达情况,分析其可能的作用机制,为NAFLD等代谢性疾病的防治提供依据。
1 材料与方法
1.1 实验动物遗传背景为C57BL/6J的雄性野生型小鼠(WT组)和PPARα基因敲除小鼠(PPARα KO组),各8只,均购置于上海南方模式生物科技股份有限公司,许可证号码:SCXK(沪)2017-0010。饲养于新疆医科大学动物实验中心SPF级屏障系统中,环境温度控制在22~25℃,相对湿度达40%~70%,光暗周期12 h,空气洁净度达1万级,自由饮水和进食。严格遵守动物伦理和福利的要求进行所有实验操作,过程均符合标准。
1.2 主要试剂及仪器基因组DNA提取试剂盒(北京天根生化科技有限公司,货号DP304-03)。总胆固醇(TC,日本和光纯药,货号635-50981);甘油三酯(TG,日本和光纯药,货号632-50991);0.5%油红O(Sigma公司,批号O0625);HE染液(Biosharp公司,货号BL700B);PPARα抗体(Cayman,货号Cay101710-1);脂滴包被蛋白2(Plin2)抗体(Proteintech公司,货号15294-1-AP);花生四烯酸ω羟化酶(Cyp4a14)抗体(Santa Cruz公司,货号SC-271983);Hsp90(货号ab13492)、脂肪酸转位酶(CD36)抗体(货号ab252922)、HRP标记的羊抗小鼠(货号ab6789)以及羊抗兔抗体IgG(货号ab6721)均购自Abcam公司。冰冻切片机、石蜡包埋和切片机(德国Leica公司);低温冷冻高速离心机(美国Thermo Scientific公司);冷冻研磨仪(浙江美壁仪器有限公司);凝胶成像分析仪ChemiDoc M、凝胶电泳仪以及PCR扩增仪(美国Bio-Rad公司);琼脂糖凝胶电泳仪(北京市六一仪器厂)。
1.3 PPARα基因敲除小鼠鉴定小鼠出生至2月龄后,剪取鼠尾约0.5 cm,放置入1.5 mL无菌EP管中,参照天根基因组DNA提取试剂盒操作方法,加入裂解液和蛋白酶K,55℃水浴过夜裂解,95℃变性后,12 000 r/min离心5 min,取上清为模板行PCR琼脂糖凝胶电泳检测。PCR反应体系(20 μL/管):2×Taq PCR MasterMix II 10 μL;引物P1、P2各0.5 μL;超纯水7 μL;DNA模板2 μL。引物P1:CTGGTCTTAAGCTCACAATCCTC;引物P2: CTCTACATGGTCACCTCTGCTCTA。PCR循环条件为:95℃ 5 min;95℃ 15 s;58℃ 15 s;72℃ 1 min;72℃ 5 min;4℃保持,2~4步循环35次,扩增结束的DNA扩增产物经凝胶电泳鉴定,并进行双向测序验证。
1.4 血清及肝组织样本收集小鼠饲养16周,两组小鼠禁食不禁水12 h后称重,腹腔注射1%戊巴比妥钠麻醉小鼠,心脏穿刺采血,血样室温放置1 h后,4℃冰箱放置2 h,4℃条件下以3 000 r/min离心10 min,收集血清样本冻存。肝脏经冰盐水漂洗后大体形态拍照,称量肝脏湿重,并将肝脏切成数个小块,液氮快速冷冻后放置-80℃冰箱,部分肝组织制成石蜡切片和冰冻切片备用。肝脏指数的计算公式:肝脏湿重/小鼠空腹体重×100%。
1.5 肝脏组织病理学分析肝脏组织经多聚甲醛溶液固定、脱水、石蜡包埋,5 μm厚切片。常规HE染色,在显微镜下观察肝组织病理结构变化。新鲜配置0.3%油红O工作液,37℃加热10 min,0.45 μm滤器过滤待用。从-80℃冰箱中取出包埋的肝脏组织,切成厚度为8 μm的冰冻切片,滴加油红O染色孵育5 min,充分水洗,水溶性封片剂封片,显微镜下每个切片随机选10个视野进行拍照,观察肝组织中脂质蓄积情况。
1.6 肝脏及血清中TC和TG含量检测取20 mg左右肝脏组织,加入1.2 mL甲醇/氯仿(1∶2体积)混合液匀浆,37℃300 r/min翻转抽提3 h,2 000 r/min离心10 min,收集上清液,加入400 μL蒸馏水震荡混匀,12 000 r/min离心10 min,收集有机溶剂层,取20 μL蒸干后加入等体积无水乙醇溶解脂质,根据试剂盒说明书测定肝脏及血清中甘油三酯和胆固醇的含量。
1.7 肝脏组织中PPARα、CD36、Cyp4a14和Plin2蛋白表达称取20 mg肝组织,加入300 μL RIPA裂解工作液并组织匀浆,经超声破碎后提取WT和PPARα KO小鼠的肝组织总蛋白,用BCA蛋白定量分析试剂盒定量后行SDS-PAGE电泳,每孔上样40 μg总蛋白。以80 V恒压电泳30 min,待样品跑过浓缩胶后,再以120 V恒压电泳1 h,并以80 V恒压3 h转至PVDF膜。经5%脱脂奶粉室温封闭2 h后,分别与PPARα、CD36、Cyp4a14和Plin2及Hsp90一抗(1∶1 000)4℃孵育过夜,TBST洗膜5次,每次5 min,后用HRP标记的Ⅱ抗(1∶1 000)室温孵育2 h,同上洗膜,ECL发光液孵育后,取出PVDF膜于凝胶成像分析仪上进行曝光分析,以Hsp90为内参照,Image Lab对目的蛋白与Hsp90灰度的比值进行蛋白表达分析。
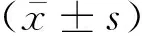
2 结果
2.1 PPARα基因敲除小鼠的构建提取小鼠的鼠尾总DNA并进行PCR扩增,PCR产物经琼脂糖凝胶电泳,检测结果显示:扩增出919 bp条带的是WT小鼠,扩增出559 bp条带的是PPARα基因敲除小鼠(如图1A),PCR产物经双向基因测序结果显示,与WT小鼠相比,PPARα KO小鼠的基因在338 bp和697 bp之间发生了缺失突变。
注:A是PPARα KO小鼠PCR鉴定,B是PPARα KO小鼠蛋白表达鉴定。图1 PPARα 基因敲除小鼠鉴定
提取小鼠肝脏组织总蛋白,Western Blot检测结果显示,在WT小鼠肝脏中检测到PPARα蛋白表达,而PPARα KO小鼠肝脏中未检测到PPARα蛋白(图1B),说明敲除PPARα基因的exon4后,导致PPARα蛋白读码框发生移码,提前终止翻译,正常的PPARα蛋白无法表达,证明成功构建获得PPARα基因敲除小鼠。
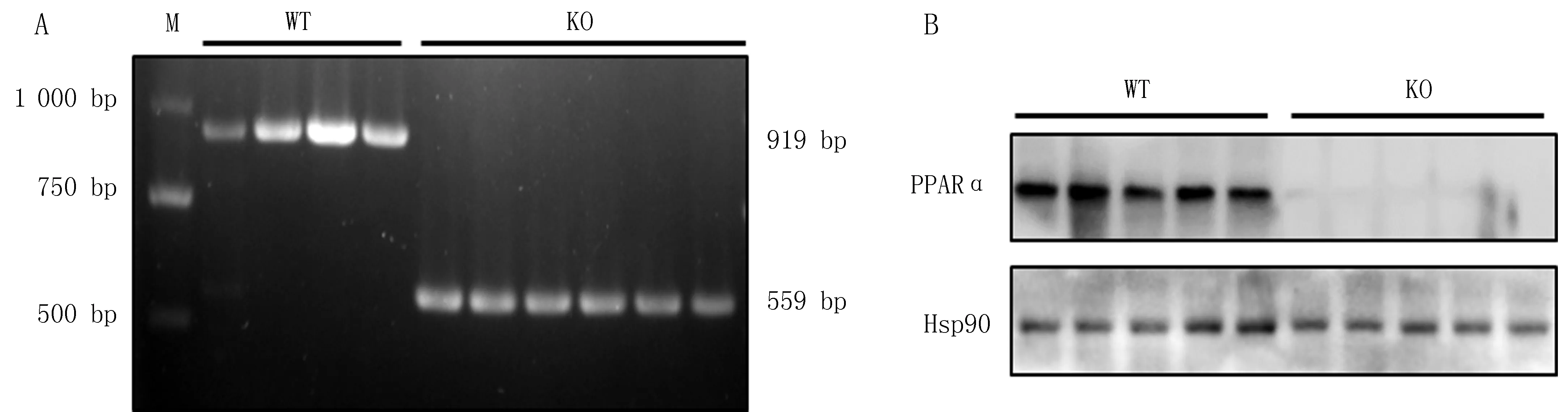
2.2 两组小鼠肝脏形态、体重、肝脏湿重和肝脏指数比较肝脏形态如图2A所示,与WT小鼠相比,4月龄的PPARα KO小鼠肝脏体积明显增大,整体呈土黄色,表面较油腻,边缘圆顿。两组小鼠体重相比差异无统计学意义[(23.30±0.44) gvs(23.11±0.69)g,P>0.05,见图2B]。与WT小鼠的肝脏湿重(0.79±0.02) g和肝脏指数(3.40±0.07)%相比,PPARα KO小鼠的肝脏湿重(1.04±0.03)g和肝脏指数(4.51±0.07)%均显著增加,差异有统计学意义(P均<0.01),见图2C、2D。
注:A是肝脏大体形态;B,C,D分别是两组小鼠体重、肝脏湿重及肝脏指数情况; n=8,与WT小鼠比较, *P<0.01。图2 两组小鼠肝脏形态、体重、肝脏湿重和肝脏指数比较
2.3 PPARα敲除小鼠肝脏组织病理学改变HE染色结果显示,4月龄PPARα KO小鼠肝细胞出现自发性肿胀,细胞形态不规则,排列结构无序,细胞内存在明显的脂滴空泡,而WT小鼠染色结果正常(图3A)。油红O染色结果显示,PPARα基因敲除后胞浆间隙内分布大小不等的脂滴,脂质沉积程度明显增加(图3B),表明PPARα基因敲除会增加小鼠肝脏的脂质蓄积。
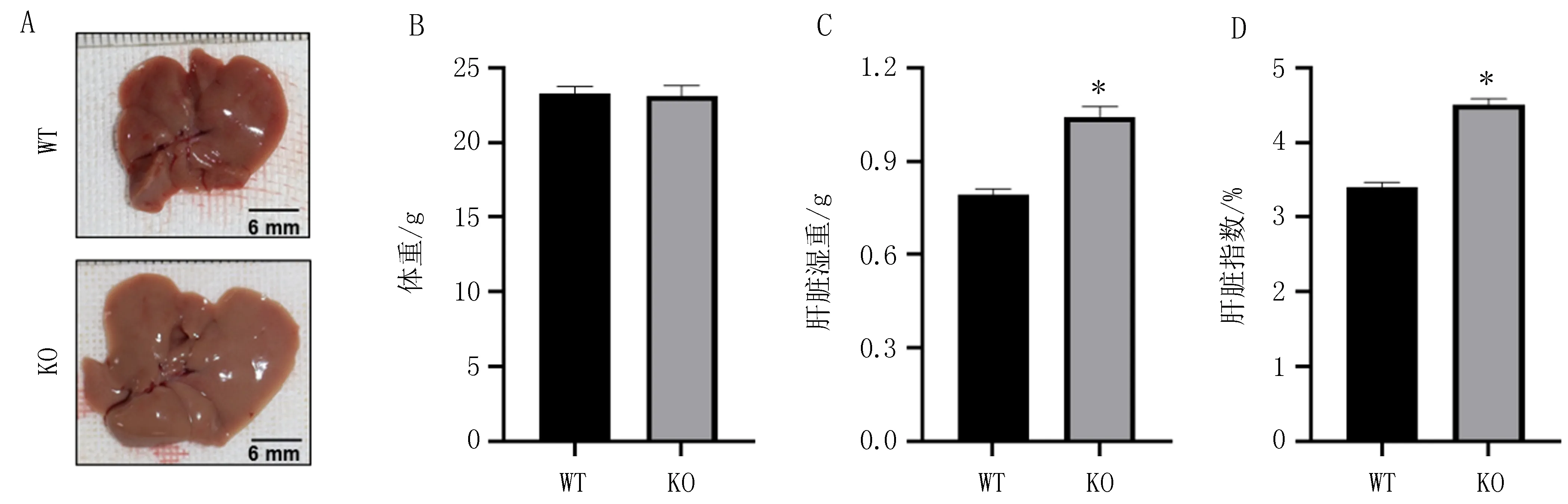
2.4 PPARα敲除对小鼠肝脏及血清TG和TC水平的影响与WT小鼠[肝脏TG含量(17.45±1.70)mg/g,血清TG水平(70.46±3.56)mg/dL]相比,PPARα KO小鼠的肝脏TG含量(33.26±3.05)mg/g和血清TG水平(122.7±14.61)mg/dL均显著增高,差异有统计学意义(P均<0.01),见图4A、4C。WT小鼠和PPARα KO小鼠肝脏TC含量[(4.41±0.21)mg/gvs(4.46±0.18)mg/g]、血清TC水平[(81.49±4.61) mg/dLvs(82.97±3.57)mg/dL]比较,差异无统计学意义(P>0.05),见图4B、4D。
注: A, B是肝脏组织中TG和TC含量; C, D是血清中TG和TC水平; n=8, 与WT小鼠比较, *P<0.01。图4 两组小鼠肝脏及血清中TG和TC水平比较
2.5 PPARα敲除对肝脏CD36、Cyp4a14及Plin2蛋白表达的影响Western Blot检测结果发现,与WT小鼠比较,PPARα KO小鼠肝脏中PPARα所调控的靶蛋白CD36和Cyp4a14的表达均显著降低,脂滴包被蛋白Plin2的表达显著增高,差异均有统计学意义(P<0.01),见表1、图5。
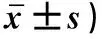
表1 两组小鼠肝组织CD36、Cyp4a14和Plin2蛋白相对表达水平比较(n=6/组,
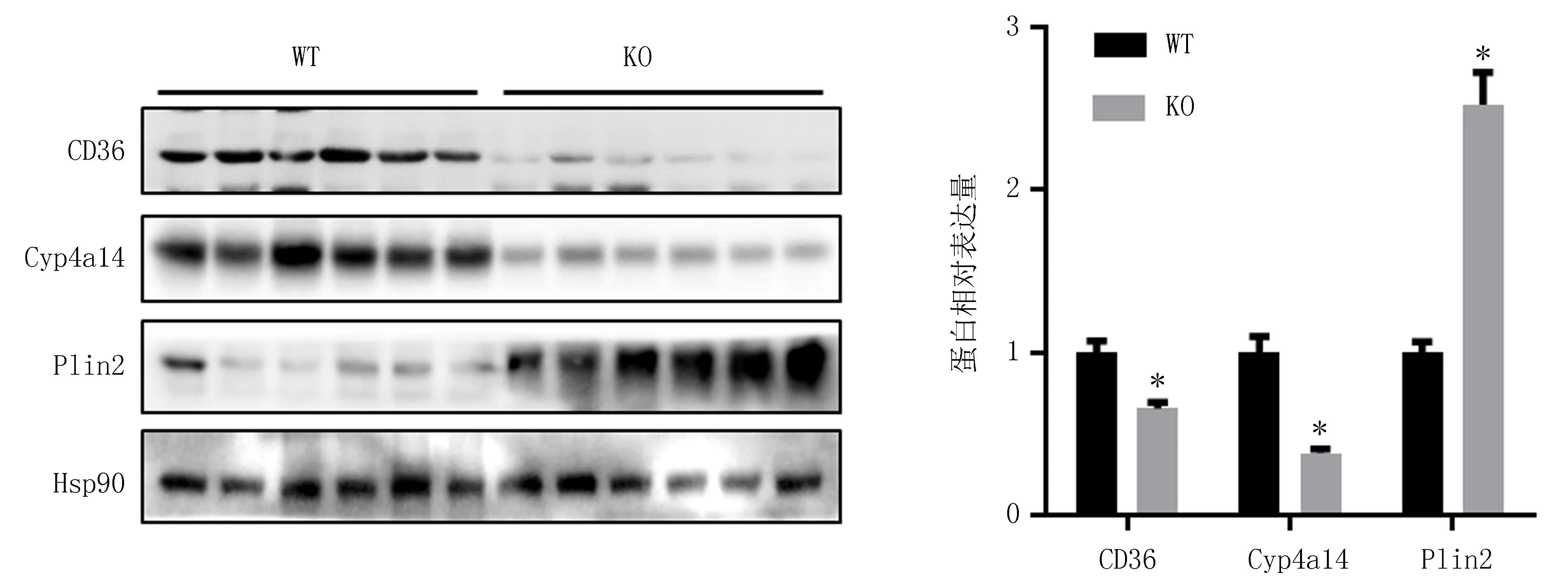
注:与WT小鼠比较, *P<0.01。图5 两组小鼠肝脏CD36、Cyp4a14及Plin2蛋白表达水平比较
3 讨论
随着人们生活方式和营养结构的改变,非酒精性脂肪肝的患病率增高,已超过病毒性肝炎居慢性肝病之首,成为威胁国民健康的重大公共健康问题[9]。NAFLD的特征是肝脏中脂肪沉积过多伴部分组织损伤或炎症,脂质代谢稳态失衡是造成肝组织脂肪变性的直接原因,导致过多的脂类物质在肝内沉积,形成NAFLD。目前,NAFLD确切的发病机制仍不明确。根据“二次打击”学说,肝脏脂肪的积累是第一次打击,使肝脏脂质代谢紊乱,脂质过量沉积的肝细胞发生氧化应激和脂质过氧化(第二次打击)从而导致肝脏损伤、炎症坏死和纤维化。因此,探寻抑制肝脏脂质积累的潜在干预治疗靶标对防控NAFLD具有重要意义。
代谢性核受体PPARα作为脂肪酸感受器和调节脂代谢的关键转录因子,与视黄醛X受体形成异二聚体复合物,移位到细胞核,与过氧化物酶体增殖物激活受体反应元件结合而促进其下游基因的转录,活化调控众多涉及脂质合成与氧化、葡萄糖摄取、炎症及免疫相关基因的表达[10]。PPARα主要通过激活氧化磷酸化和脂肪酸β氧化相关通路来降低肝脏脂质水平,此外,还参与调控多条通路,如过氧化体相关的氧化脂肪酸、乙酰辅酶A羧化酶、核受体基因等发挥调节脂质代谢和能量代谢的功能[11]。既往研究表明肝细胞特异性的PPARα缺失可诱导衰老小鼠(51周)自发性的肝脏脂肪变性[12],高脂饮食会诱发全身性及肝脏特异性PPARα基因敲除的小鼠发生严重的脂肪肝[13]。PPARα基因敲除小鼠肝脏中游离脂肪酸含量明显增多,脂质代谢明显紊乱[14]。本研究结果显示,PPARα基因敲除后导致小鼠肝脏内脂肪沉积增多出现明显的脂肪变,同时显著升高血清TG浓度,提示4月龄PPARα基因敲除小鼠在普食喂养条件下可自发出现明显的肝脏脂肪变性和高甘油三酯血症表型,表明PPARα对维持肝脏脂质稳态至关重要,PPARα参与了肝脏脂肪代谢。因此PPARα可能成为防治NAFLD的潜在药物干预靶标[15-16]。
研究表明PPARα激活剂活化PPARα后可通过诱导下游脂肪酸转运体CD36以及脂肪酸氧化酶Cyp4a14等靶基因高水平转录,促进脂肪酸在过氧化物酶体和线粒体中的分解和氧化[17]。PPARα可通过增加Cyp4a的表达,催化不饱和脂肪酸在ω和ω1位点羟基化,加速脂肪酸氧化。在本研究中,PPARα基因敲除小鼠肝脏CD36和Cyp4a14蛋白表达显著降低,而脂滴包被蛋白Plin2表达水平显著增高,推测可能由于敲除了PPARα,CD36的表达降低,肝细胞内脂肪酸向内质网和线粒体膜的二次转运受抑制,从而过多的脂肪酸聚集在胞内[18],同时Cyp4a14的表达也降低,抑制了脂肪酸的氧化,而脂滴包被蛋白Plin2的表达增高促进了脂滴吸收和脂质堆积。
综上所述,本研究通过普通饮食喂养WT和PPARα KO小鼠,发现4月龄PPARα KO小鼠会出现肝脏肿大、肝脏脂质堆积和高甘油三酯血症等NAFLD的表型,为以PPARα为靶点治疗非酒精性脂肪肝等代谢性疾病的治疗药物研发提供实验依据。
