对乙基苯甲醚的电化学苄位唑胺化反应的探索
2023-11-02胡新伟阮志雄
胡新伟,阮志雄
广州医科大学药学院,广州 511436
1 引言
电化学合成一般在电流条件下促使反应物得到或失去电子,发生后续的氧化或还原反应。而且在反应过程中无需添加氧化剂或还原剂,反应物可以直接提供或得到电子,整个反应体系符合绿色化学的要求[1-3],因此近年来备受关注。其优点可概括为:反应只需常温、常压等温和条件;通过电极进行电子转移避免了有毒或危险还原剂和氧化剂的使用;产品后处理简单且易分离,对环境污染小;利用电流强度和电解时间控制反应速率以此实现电子转移和化学反应同时进行,减少了副反应的发生。我们在本科创新实验中引入电合成方法,期望有助于学生关注学科前沿,了解绿色有机合成与发展的新趋势。
N-杂环化合物是天然产物和药物分子的重要骨架,广泛存在于自然界中,发展高效制备N-杂环化合物的有机合成方法学也逐渐成为有机合成化学的研究热点之一。早期合成N-杂环的方法多为唑类化合物与缺电子的卤代芳烃反应,在过量强碱的作用下发生亲核取代反应构建C―N键[4]。随着过渡金属催化的快速发展,Lundgren教授团队[5]报道了Cu(I)和过量MnO2催化关于α-羧酸酯-芳基苄与吡唑的偶联反应。近几年,吴骊珠教授[6]、孙江涛教授[7]、朱戎教授[8]在光催化下分别利用肟酯、重氮、烯烃与唑类化合物反应生成苄基唑类化合物,使得N-杂环化合物的合成方法学进入了一个新阶段(图1)。然而这些反应会使用过量强碱、氧化剂、催化剂,甚至是在低温条件下才能控制反应的发生,增加了反应的操作难度。虽然近年来许多课题组探索了合成唑胺化产物的新方法,但合成这类重要化合物的绿色方法仍相对较少,因此,我们依然需要开发这类重要N杂环化合物的绿色合成方法。不仅如此,我们发现四氮唑类化合物广泛应用在抗真菌类药物中,其合成途径相对复杂,而且反应条件比较苛刻,不利于最终氮唑类药物的合成。而我们发展的电合成方法可以一步生成α-羰基四唑化合物,为构筑复杂结构的四氮唑类药物提供了关键中间体。
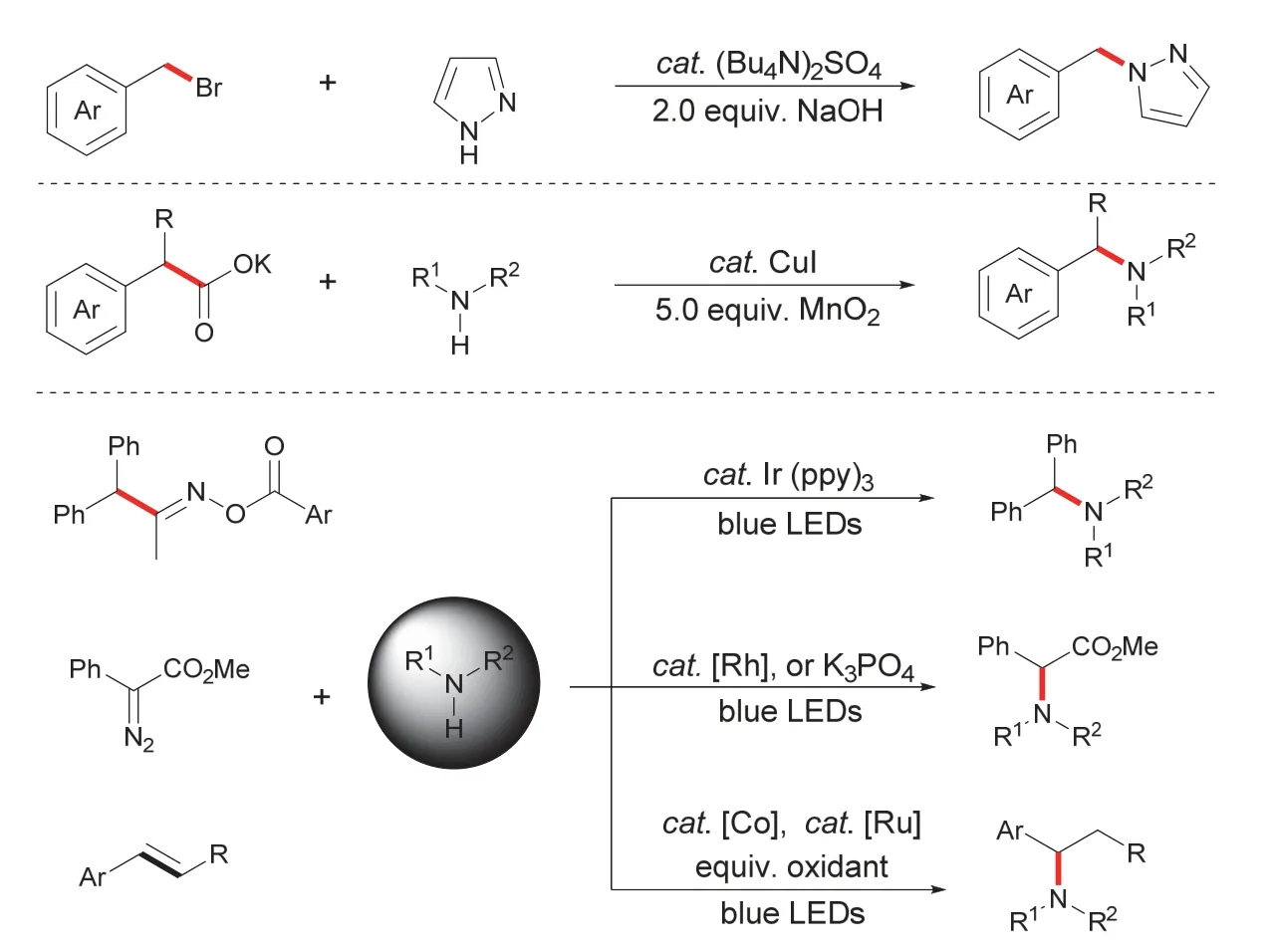
图1 传统的苄位唑胺化反应示例
在目前国内外实验教材中,均未涉及有机电合成反应。但随着近几年有机电化学的发展,一些优秀的研究成果开始应用于本科教学的探索中[9]。鉴于氮杂环结构被广泛用于天然产物和药物分子以及电化学的飞速发展,我们想是否可以将本课题组前期发展的电化学苄位唑胺化反应研究成果[10],通过改进实验方案,缩短反应时间转化为适用于本科教学的有机创新综合实验(图2)。因此,我们选择对乙基苯甲醚和5-苯基四氮唑为底物,利用简单的反应装置,一步进行C―N键构建来合成苄位胺化芳烃。这些创新实验可以让学生在实验中学会使用薄层色谱(TLC)监测反应,柱层析技术分离产物以及利用核磁共振和高分辨质谱手段对产物进行表征分析。与传统的有机合成方法不同的是,开展电化学相关的创新实验是对重要有机分子的高效合成训练,不仅有助于启发学生对绿色有机合成的新思考,而且在训练学生综合运用有机合成知识开发新反应以及指引学生关注碳中和远景方面起到了积极的作用。

图2 电化学苄位C―H唑胺化反应
2 实验部分
2.1 循环伏安实验
为了更好地引导学生思考该反应的原理,我们测试了原料1和2的氧化还原电势(图3),发现它们的氧化电位分别为1.98 V (vs.Ag/AgCl)和2.00 V (vs.Ag/AgCl),产物3的氧化电位2.14 V (vs.Ag/AgCl),比原料1和2都高。以上结果说明原料1最容易被氧化,失去电子,而且其经历两次氧化,最后得到产物3。
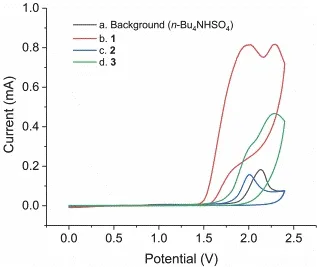
图3 原料及产物的循环伏安图
2.2 实验原理
电化学有机合成反应可以直接在电极表面发生,也可以通过间接电解,引入氧化还原媒介,进而在溶液中发生反应。本实验采用直接电解的方法,在电极表面发生反应。基于前期研究[10,11],反应机理如图4所示:该反应首先由石墨阳极表面发生氧化开始,芳环失去电子产生自由基阳离子A,随后通过电子迁移生成苄基自由基B。B进一步被阳极氧化为苄基正离子C,然后与5-苯基四氮唑发生亲核取代反应,离去一个质子,生成最终唑胺化产物3。同时,质子经过铂阴极表面还原生成H2,从而消除了对牺牲化学氧化剂的需求。
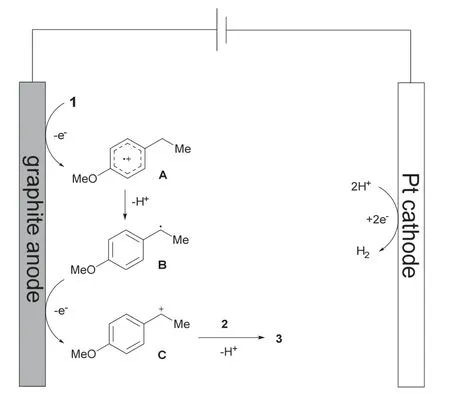
图4 可能的反应原理
2.3 实验方案的优化
原实验方案中,反应温度需要80 °C,时间需要12 h,难以满足本科教学需求。本文对反应条件进行了改进,通过探究不同条件(温度、电流、时间)对该反应的影响(表1),以期望将反应条件优化至合理范围。首先控制温度在25 °C,通过改变电流和反应时间发现产率最高为74%,可能的原因是温度太低导致化合物活性不高,反应相对缓慢。而当增加温度至50 °C时,增加反应电流至16 mA,反应6 h能得到最高产率81%。继续升高温度至80 °C,产率没有较高提升。相较而言,反应中使用的溶剂为乙腈,80 °C下接近沸腾,不方便操作。使用其他相对便宜的石墨片、铁片和镍片作为阴极时,收率相对标准条件下稍逊。综合考虑后,我们选择石墨片(1.0 cm × 1.0 cm × 0.2 cm)作为阳极,铂片(1.0 cm × 1.0 cm × 0.01 cm)作为阴极,四丁基硫酸氢铵作为电解质,乙腈作为溶剂,反应温度50 °C、电流16 mA、反应时间6 h为标准反应条件。
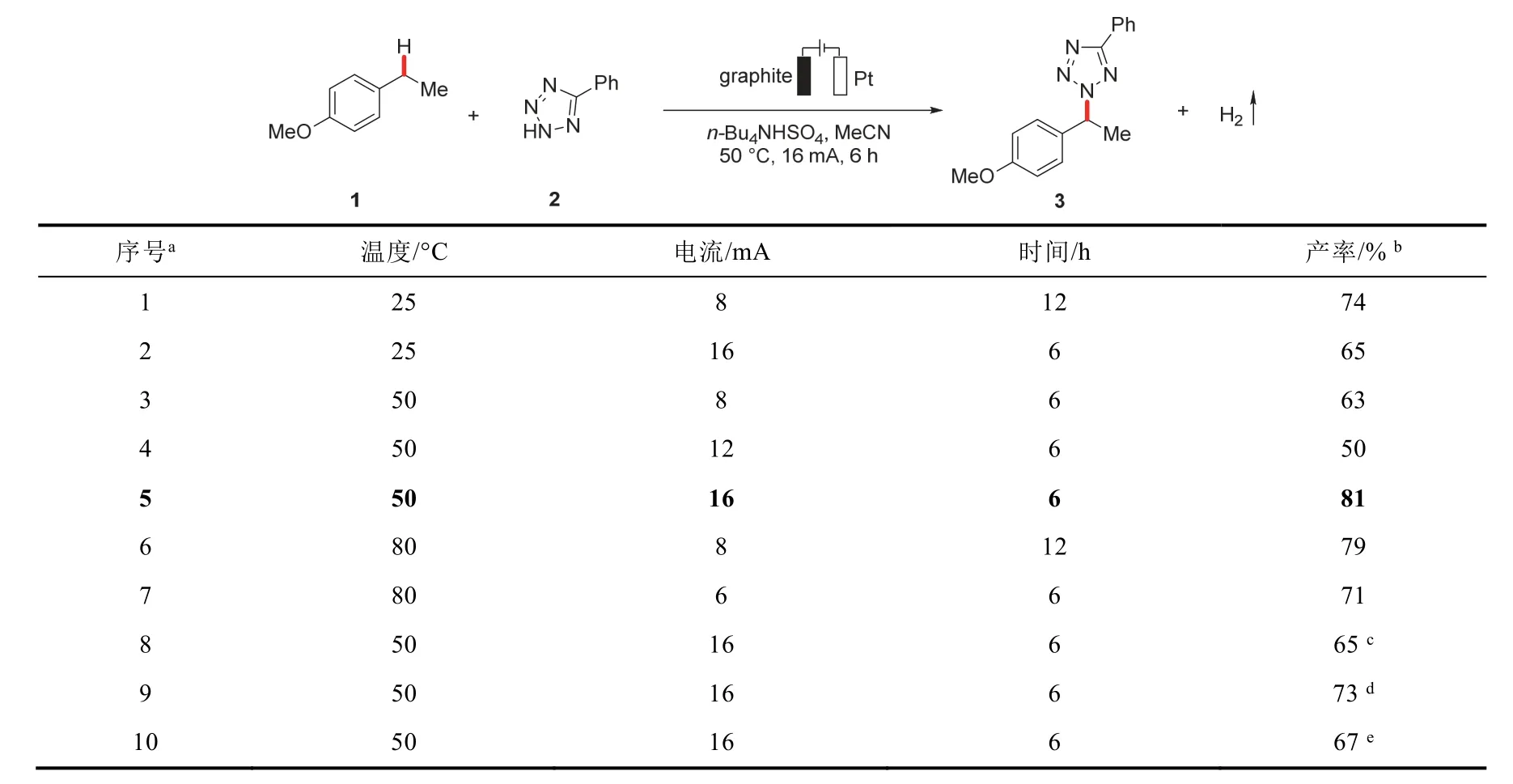
表1 不同反应条件对反应进程的影响及产物收率比较
2.4 反应的可重复性
筛选出最优的反应条件后,我们对标准底物反应进行了多次重复,并最终确定该电化学反应具有较好的重现性。实验选取对乙基苯甲醚和5-苯基四氮唑为原料,以铂片和石墨片为阴阳极,三名学生(A,B,C)分别进行2次实验,综合评价反应过程和效果(表2)。反应过程中使用薄层色谱板监测(展开剂:V石油醚(PE):V乙酸乙酯(EA)= 10 : 1),现象基本一致,产率在误差范围内能够很好地重复,有助于推广于本科创新实验。

表2 优选条件的复现性实验
2.5 趣味性实验
为增加反应的趣味性,加深同学们对反应的理解,我们将实验室中的小实验和日常生活联系起来,用生活中常见的物品代替实验室用品,真正实现科研走进生活。在分析完该反应使用的电极后,我们设想是否可以选择铅笔头作为阳极替换石墨片,阴极仍然以铂片为电极,来尝试该反应。
实验依然是以对乙基苯甲醚和5-苯基四氮唑为原料。准备一支削好的2B铅笔代替石墨电极(图5),依次称量对乙基苯甲醚(204 mg)、5-苯基四氮唑(73 mg)和四丁基硫酸氢铵(169 mg),并用移液枪吸取5 mL乙腈于电化学平底反应瓶中,将电化学平底反应瓶置于磁力搅拌器中心,启动磁力搅拌器。三名学生(A,B,C)分别进行实验,调节电流为16 mA,反应液在50 °C下搅拌6 h,过程中观察到反应液颜色由无色变为淡黄色,最后变为棕黄色。而且反应过程中使用薄层色谱(TLC)监测反应进程。具体反应情况如下:
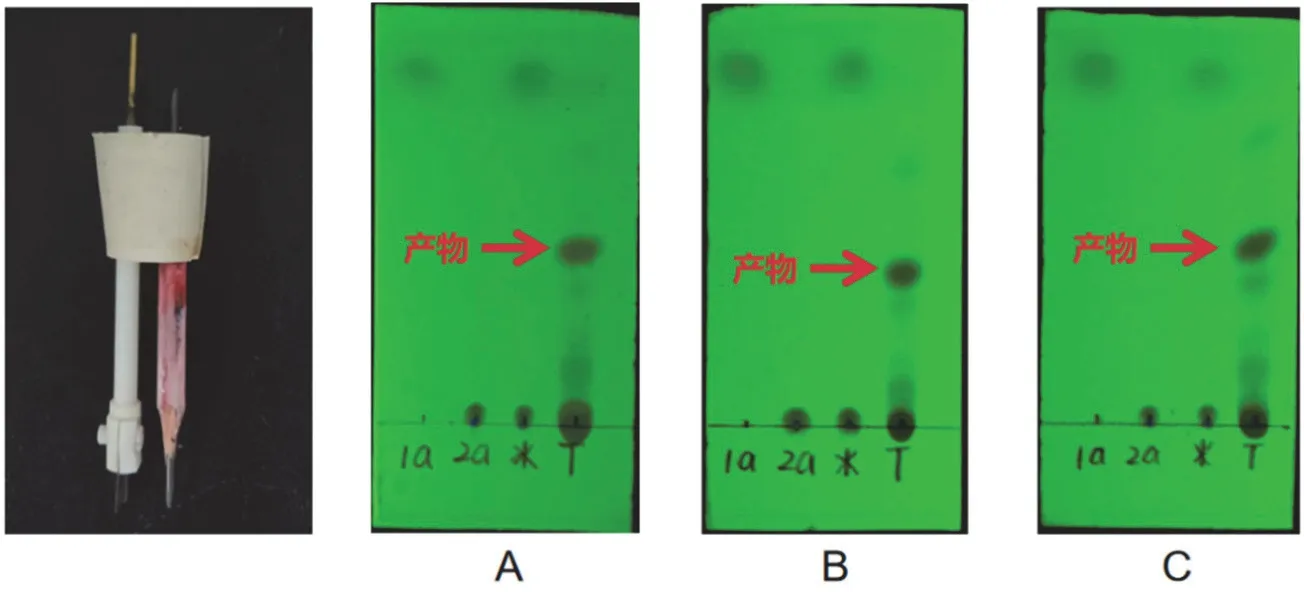
图5 铅笔电极及A、B、C三位同学的薄层层析色谱(TLC)结果
进一步将反应液转移至圆底烧瓶中,真空下旋干,选择乙酸乙酯和石油醚(VPE:VEA= 20 : 1)为洗脱液过柱分离,收集产物后称重,得到淡黄色液体,三位同学的反应收率分别为:60%,62%,66%(表3)。表明该反应有很好的重现性,即使用表面积小的铅笔芯作为阳极电极进行反应,依然可以取得可观的收率。
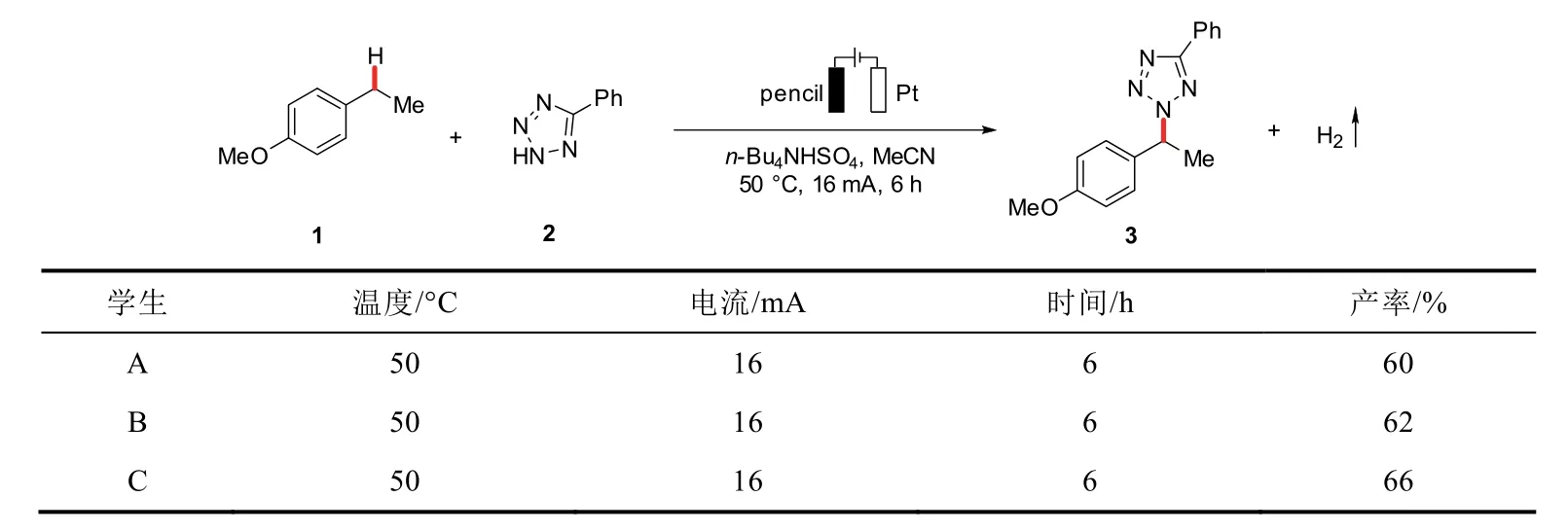
表3 趣味性实验的复现
2.6 试剂与仪器
2.6.1 试剂耗材
对乙基苯甲醚、5-苯基四氮唑、四丁基硫酸氢铵、乙腈、二氯甲烷、无水硫酸镁、氘代氯仿(98%D)(以上试剂为分析纯,购自安耐吉化学、上海毕得医药等试剂公司);石油醚(60-90 °C)、乙酸乙酯(以上溶剂为分析纯,购自广州佳钻生物科技有限公司);柱层析硅胶(300-400目,山西诺泰);石墨片(1.0 cm × 1.0 cm × 0.2 cm,北京晶龙特碳有限公司),铂片(规格为1.0 cm × 1.0 cm × 0.01 cm,天津艾达),铅笔芯(自备)。
2.6.2 仪器
400 M核磁共振仪NMR (日本JEOL)、高分辨质谱仪HR-MS (日本JEOL)、MPD-3003S直流稳压电源(伊利诺斯工具制品)、B13-3型恒温定时磁力搅拌器(上海司乐)、RV10型旋转蒸发仪(德国IKA)、分析天平(德国赛多利斯)、WFH-204BS手提式紫外分析仪(杭州齐威仪器)、移液枪(美国Thermo)、真空系统、圆底烧瓶、10 mL平底反应瓶、分液漏斗、锥形瓶、电极夹、磁子、注射器、反口胶塞、针头等。
2.7 表征方法
取30.0 mg左右的样品溶于0.6 mL氘代氯仿(CDCl3)中,转移至核磁管中充分摇匀,室温下利用核磁测定其氢谱(1H NMR)和碳谱(13C NMR)。再用高分辨质谱仪(HR-MS)测定其分子量,进一步分析确定结构。
2.8 实验步骤
1) 苄位唑胺化产物的制备。
取干燥的电化学平底反应瓶,装入洁净搅拌子。分别称取对乙基苯甲醚(204 mg,1.5 mmol),5-苯基四氮唑(73 mg,0.5 mmol)和四丁基硫酸氢铵(170 mg,0.5 mmol),并迅速转移至反应瓶内。用移液枪吸取5 mL乙腈于反应瓶中,然后将带有收集尾气的气球以及铂片电极(1.0 cm × 1.0 cm × 0.01 mm)和石墨电极(1.0 cm × 1.0 cm × 2 mm)的橡胶塞插入反应瓶,再将反应瓶置于磁力搅拌器中进行搅拌,并将温度设为50 °C。将铂电极与直流稳压电源的负极相接,石墨电极与电源的正极相接,启动直流稳压电源,调节电流稳定至16 mA,反应6 h。反应过程中可观察到反应液由无色变为棕黄色(图6)。

图6 实验涉及的反应装置
2) 反应进度监测。
反应过程中,每隔2 h用毛细管吸取少量反应液进行点板,并与原料点及混合点对比。以VPE:VEA=10 : 1为展开剂做TLC分析(图7),监测反应进度,直至原料2a反应完全停止反应。

图7 不同反应时间的薄层层析色谱结果
3) 柱层析分析。
用二氯甲烷将电极上附有的反应液洗至电化学平底反应瓶内,再用一次性吸管将反应瓶中的反应液转移至茄型瓶中,用二氯甲烷润洗三次。往茄型瓶中加入2勺100-200目硅胶粉,用旋转蒸发仪将粗产物旋干,得到吸附有粗产物的黄棕色硅胶。
固定带有砂芯的色谱柱,依次填装300-400目硅胶、少量石英砂、粗产物、石英砂,选用乙酸乙酯和石油醚的混合液为洗脱液过柱,洗脱液的用量及比例依次为50 mL的纯石油醚、450 mL的VPE:VEA= 20 : 1,用试管收集需要的产物点,并通过TLC时刻关注流出液情况,防止过漏产物,确定产物分布。收集含有产物的流出液于圆底烧瓶中,用旋转蒸发仪旋干浓缩,得到淡黄色粘稠状液体。取一个25 mL的圆底烧瓶称重后,用二氯甲烷将产物转移至烧瓶中,真空下旋干溶剂,抽真空15 min,称量最后的瓶重,得到淡黄色黏性液体114 mg,收率81%。转入2 mL塑料离心管中,贴好标签保存。
3 结果与讨论
3.1 产物的表征
对苄位唑胺化产物分别进行核磁共振氢谱(1H NMR)和碳谱(13C NMR)表征(图8和图9)。
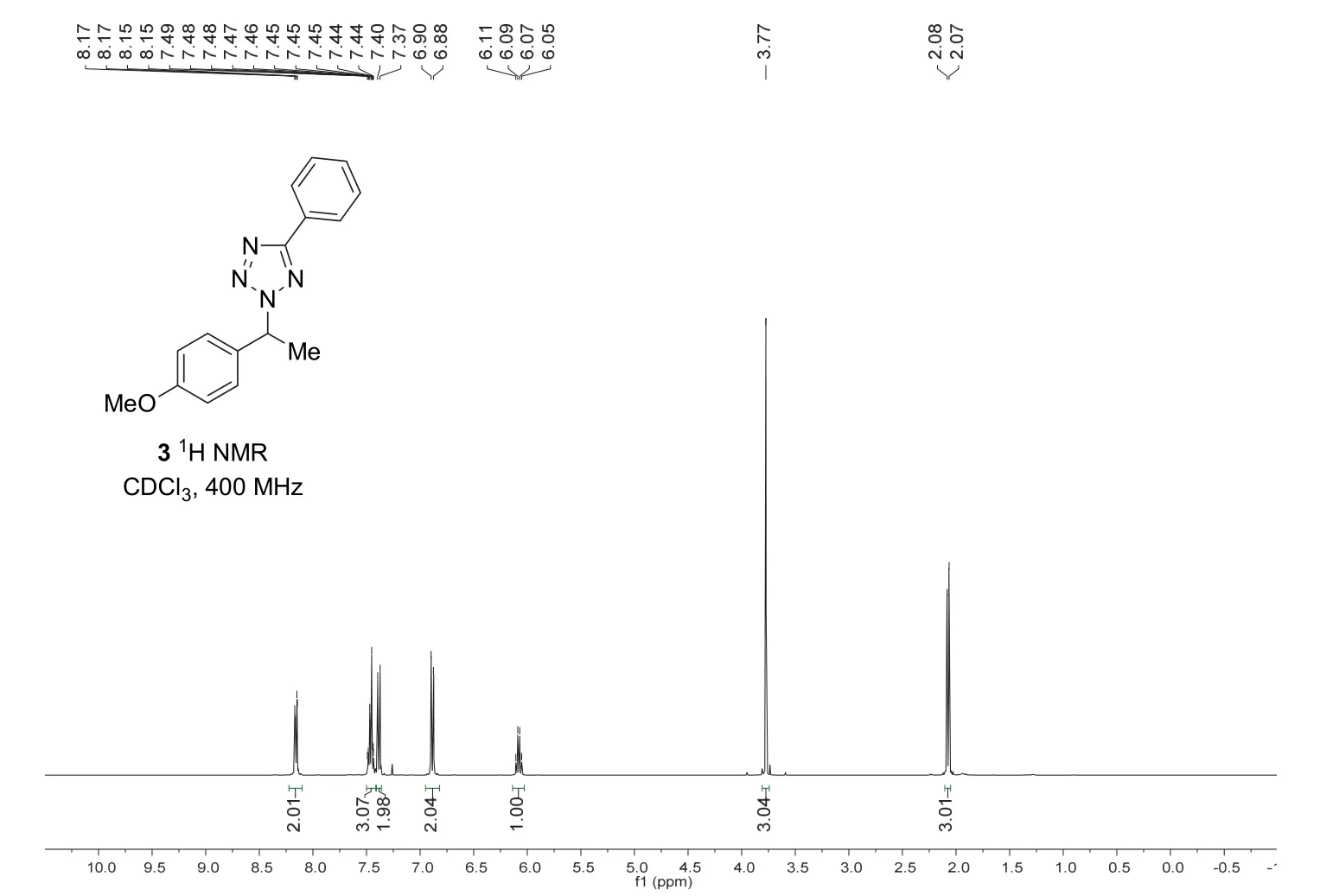
图8 苄位唑胺化产物的核磁氢谱
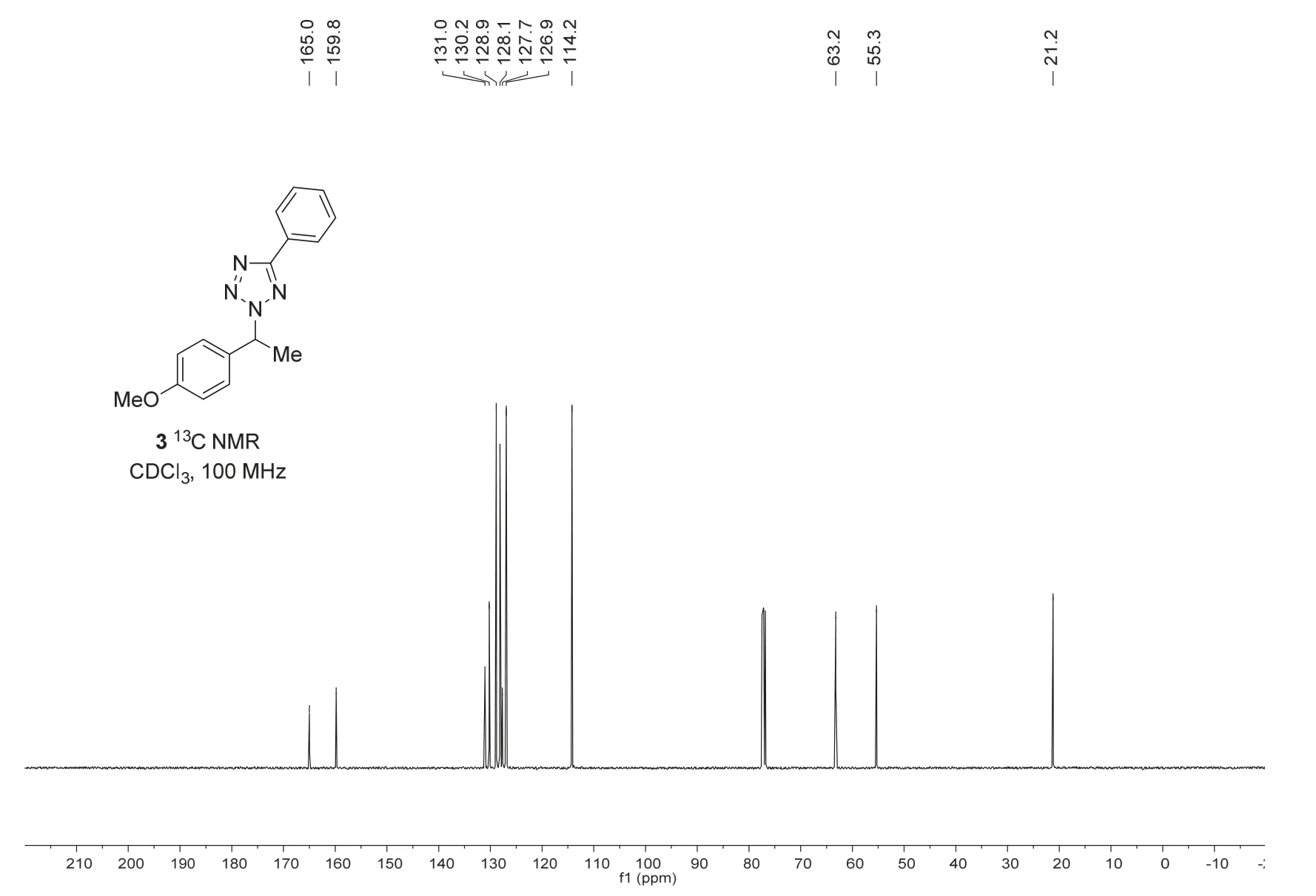
图9 苄位唑胺化产物的核磁碳谱
表征数据:1H NMR (400 MHz, CDCl3)δ8.16 (dd,J= 7.7, 1.9 Hz, 2H), 7.50-7.43 (m, 3H), 7.38 (d,J= 8.7 Hz, 2H), 6.89 (d,J= 8.7 Hz, 2H), 6.08 (q,J= 7.2 Hz, 1H), 3.77 (s, 3H), 2.08 (d,J= 7.2 Hz, 3H);13C NMR (100 MHz, CDCl3)δ165.0, 159.8, 131.0, 130.2, 128.9, 128.1, 127.7, 126.9, 114.2, 63.2, 55.3,21.2。HR-MS (ESI)m/zcalcd for C16H16N4ONa [M+Na]+303.1216, found 303.1214。
3.2 实验讨论
该创新实验利用简单绿色的电化学合成方法高效制备了苄位唑胺化产物。以石墨片为阳极,铂片为阴极,反应仅需加入一个当量电解质,在单电解槽中常温恒流(16 mA)反应,即能以81%的收率得到相应产物。反应时间可根据底物用量和恒电流强度估算,且实验现象较明显,后处理简单,反应过程涉及有机化学和物理化学交叉学科的应用。在进行本科教学实验时,可以合理预估时间,反应前的准备工作0.5 h,反应6 h,后处理需要0.5 h,分离纯化2 h,共9 h。
不仅如此,该教学实验紧密联系当下绿色化学合成热点,积极融入了以“科学问题为导向”的探究型实验方案,在引导学生关注学科前沿的同时也拓宽了自己的思维。且利用有机方法学研究的一般过程,在合成产物方面具有广泛的衍生空间,适合本科生开展不同类型或底物结构的创新实验研究。
4 结语
本文以电化学Csp3―H胺化反应构筑C―N键为背景,实现了苄位烷基与胺的脱氢偶联反应。通过将科研成果转变为本科教学实验,开发了“对乙基苯甲醚的电化学苄位唑胺化反应研究”实验。实验涉及的C―H键官能团化反应,为当下有机合成化学的前沿领域。我们尝试将国际前沿与本科实验相结合,应用有机化学知识深入探索含氮化合物的新型合成方法。以本实验方案为蓝本,增强学生对薄层色谱、柱层析纯化、核磁共振波谱分析等实验技能的理解,进一步添加趣味性实验,融入安全、可持续的绿色化学理念,综合训练学生的设计和思维,以及解决复杂合成问题的能力。
