新生儿先天性肺泡蛋白沉积症1例
2023-11-01韩俊彦张蓉周建国胡黎园钱莉玲陆爱珍杨琳马阳阳乔中伟张澜
韩俊彦 张蓉 周建国 胡黎园 钱莉玲 陆爱珍 杨琳 马阳阳 乔中伟 张澜
(1.复旦大学附属儿科医院新生儿科/国家儿童医学中心/卫生部新生儿疾病重点实验室,上海 201102;2.复旦大学附属儿科医院呼吸科/国家儿童医学中心,上海 201102;3.复旦大学附属儿科医院内分泌遗传代谢科,上海 201102;4.复旦大学附属儿科医院病理科/国家儿童医学中心,上海 201102;5.复旦大学附属儿科医院影像科/国家儿童医学中心,上海 201102)
1 前言
肺泡蛋白沉积症 (pulmonary alveolar proteinosis, PAP)是一种少见的弥漫性肺间质病变,发生率为6.9/100 万~8.0/100 万[1-2]。该病1958年由Rosen等[3]首次报道,以肺泡巨噬细胞功能障碍和肺泡表面活性物质在肺泡腔积聚,引起换气障碍,进而导致呼吸衰竭为主要临床表现。按起病原因不同,PAP 分为原发性(>90%)、继发性(5%~10%)和先天性(2%)三大类[4-5]。原发性PAP 主要是由于粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony stimulating factor,GM-CSF)信号途径异常,肺泡蛋白代谢物质清除减少、堆积增多所导致;原发性PAP 分为自身免疫性(>90%)和遗传性(编码GM-CSF 受体亚基基因CSF2RA或CSF2RB突变所致)。继发性PAP是由各种可能影响肺泡巨噬细胞数量或功能的疾病引起,如骨髓增生异常综合征、慢性炎症、恶性肿瘤、毒物暴露等。先天性PAP 多数在新生儿期或婴幼儿期起病, 主要由SFTPB、 SFTPC、ABCA3、NKX2.1等编码肺泡表面活性蛋白或参与表面活性物质分泌的基因突变引起的肺表面活性物质生成异常或功能障碍所导致[6]。新生儿期常见表现为呼吸窘迫综合征,对常规治疗无反应或仅有一过性反应。先天性PAP 整体预后较差,且预后与基因突变的类型和方式有关[7]。
2 病例介绍
现病史:患儿,男,44 日龄,因生后呼吸困难、不能离氧1月余收住复旦大学附属儿科医院新生儿重症监护病房。患儿系第3胎第3产,经阴道分娩出生,胎龄40+1周,出生体重3 700 g,出生时羊水、脐带、胎盘均未见明显异常,生后Apgar评分1 min 10分,5 min 10分。生后2 h出现呻吟、吐沫、皮肤青紫,收入当地医院新生儿科治疗44 d;出院诊断:呼吸衰竭、新生儿败血症、急性呼吸窘迫综合征、新生儿肺炎、新生儿ABO 溶血、新生儿贫血、新生儿慢性肺病、肺动脉高压。在当地医院住院期间患儿不能离氧,先后予气管插管有创呼吸支持16 d,无创呼吸支持治疗8 d,高流量鼻导管吸氧22 d,住院期间分别于生后第1天及第3天给予肺表面活性物质气管内滴入治疗。患儿肺间质病变明显,当地医院予布地奈德雾化、地塞米松及甲泼尼龙治疗。住院期间也因考虑感染先后使用亚胺培南、阿莫西林克拉维酸钾、美罗培南、替考拉宁、头孢唑肟、阿奇霉素等抗感染治疗。心脏彩超提示存在轻度肺动脉高压、三尖瓣反流、卵圆孔未闭,未予特殊治疗。住院期间曾予以丙种球蛋白、白蛋白、去白红细胞悬液等血制品支持治疗。治疗44 d 后因仍呼吸费力、离氧困难,由120转诊至我院。转运途中予高流量鼻导管吸氧[7 L/min,吸入气氧浓度(fraction of inspired oxygen, FiO2)70%],哭闹时面色青紫,血氧饱和度维持在90%左右。父母既往身体健康,母亲第1胎为足月男孩,现10岁,身体健康;第2胎为足月男孩,生后3 d因呼吸困难夭折,具体原因不明;否认家族遗传性疾病史。
入院体格检查:体温36.5℃,呼吸70 次/min,心率152 次/min,血压74/46 mmHg,头围33.5 cm,身长54 cm,体重3.52 kg(出生体重3.7 kg)。神志清楚,反应欠佳,营养状态差。高流量鼻导管吸氧下全身皮肤青紫。无皮疹,无出血点。前囟平、软,大小约1.0 cm×1.0 cm。口唇稍青紫,呼吸急促,吸凹征明显。双肺听诊呼吸音粗,可闻及少许湿啰音。心音有力,心律齐,未闻及杂音。无腹胀,肝肋下2 cm,脾肋下未及,肠鸣音5 次/min。四肢肌力、肌张力正常。神经系统体格检查未见明显异常。毛细血管再充盈时间<3 s。
入院后辅助检查:血气分析示pH 7.39~7.42(参考值:7.35~7.45),二氧化碳分压49.0~54.7 mmHg(参考值:35~45 mmHg),碱剩余7.6~8.9 mmol/L (参考值:-3~3 mmol/L),乳酸0.4~1.3 mmol/L(参考值:0.5~1.6 mmol/L);血常规、C反应蛋白、肝肾功能、凝血功能、甲状腺功能等检查均无明显异常;痰培养及宏基因组二代测序阴性;血培养及宏基因组二代测序阴性;支气管肺泡灌洗液(bronchoalveolar lavage fluid, BALF)宏基因组二代测序阴性(背景菌:假肺炎链球菌);血清弓形虫、巨细胞病毒、风疹病毒、单纯疱疹病毒IgM 阴性;血尿串联质谱检测未见明显异常。胸腹X线检查提示:双肺透亮度明显减低,心缘模糊(图1A)。胸部CT 增强提示:双肺弥漫性间质病变,呈“铺路石征”(图1B~D)。心脏彩超提示:卵圆孔未闭(2.8 mm)、轻度肺动脉高压、三尖瓣反流。腹部B超未见明显异常。头颅MR平扫未见明显异常。支气管镜检查提示:双肺支气管黏膜炎性改变。BALF 病理学检查结果:苏木精-伊红(hematoxylin-eosin, HE)染色和瑞氏染色示较多组织细胞(15~25 个/高倍镜视野),少量上皮细胞和中性白细胞;特殊染色结果:过碘酸希夫(periodic acid-Schiff, PAS)染色少量阳性,铁染色阴性。家系全外显子组检测提示:患儿检测到ABCA3基因复合杂合变异,其中错义变异(NM_001089:exon7:c.557C>T(p.P186Leu),遗传自母亲(图2);ABCA3基因包含19~21号外显区域7 kb的缺失,遗传自患儿父亲。

图1 患儿胸部影像学检查结果 A:胸部X线检查示双肺透亮度减低,支气管充气征,心缘模糊,白肺;B:胸部CT示双肺透亮度减低,未见明显气道狭窄;C~D:胸部CT示双肺弥漫性间质改变,呈“铺路石征”(箭头所示)。
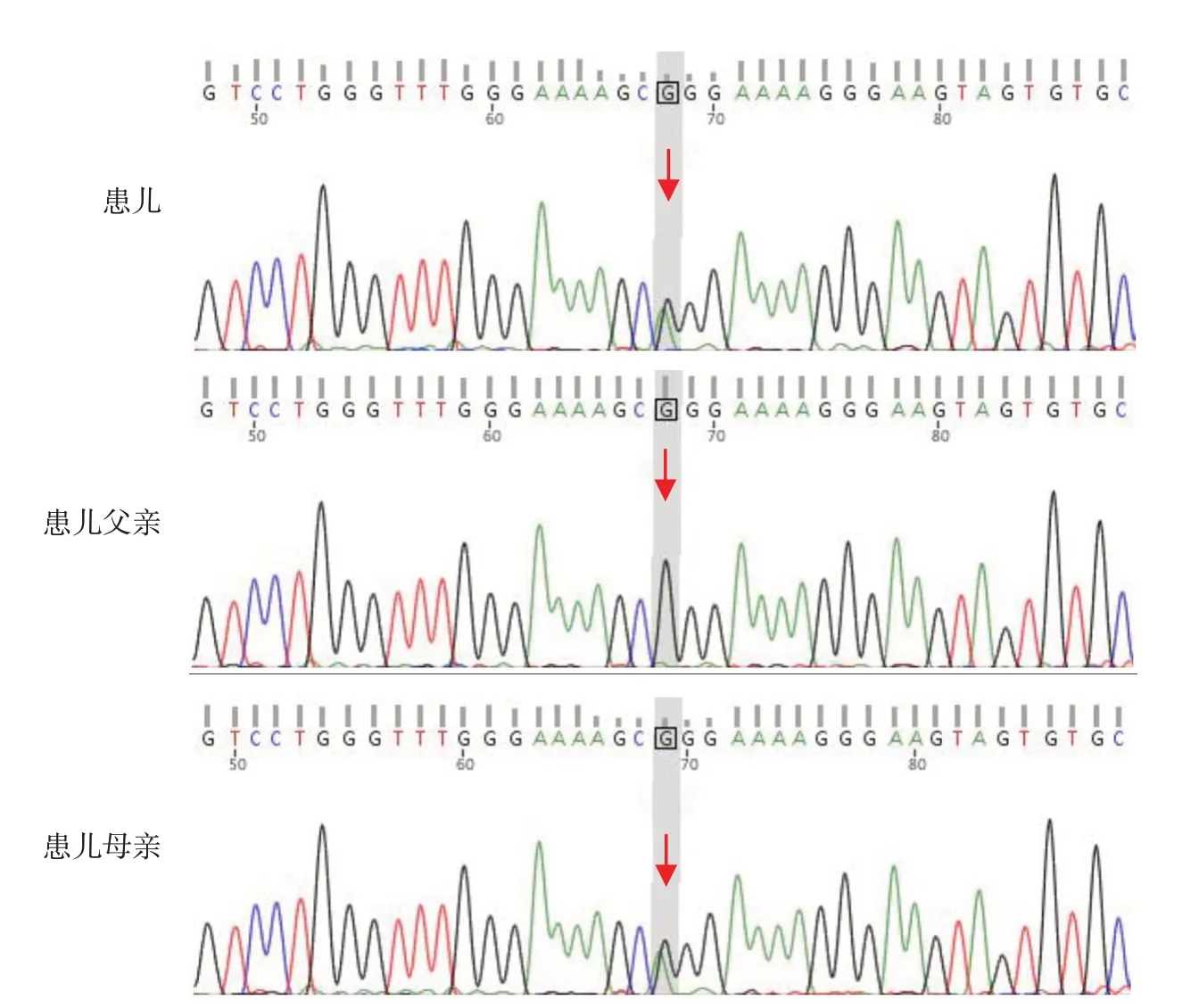
图2 患儿及其父母ABCA3 基因变异Sanger 测序图 患儿ABCA3 基因存在变异,变异位点为c.557C>T(p.P186Leu),该变异来源于母亲。箭头所示为变异位点。
3 多学科诊疗
3.1 新生儿重症监护室初诊
该患儿生后不久即出现呼吸困难,经外院氧疗、抗感染、抗炎等常规治疗后仍不能撤离有创呼吸支持,且依赖高浓度氧(FiO260%~80%),体格检查发现患儿主要表现为呼吸困难、发绀。生后长期不能撤离呼吸支持的疾病原因可分为呼吸系统疾病和非呼吸系统疾病两大类。该患儿入院后评估心血管系统功能、中枢神经系统、神经肌肉功能等均未见明显异常,故非呼吸系统疾病可能性小。呼吸系统疾病中又可分为肺实质性病变与间质性病变,首先需排除如结核分枝杆菌、巨细胞病毒等特殊病原体引起的肺部感染性疾病。因此针对上述多种病因进行初步筛查,包括血、BALF培养及宏基因组二代测序检查,完善胸部增强CT、支气管镜、BALF病理学检查等,并同时完善基因检测。
3.2 呼吸科会诊
呼吸系统疾病是导致新生儿呼吸困难的最常见病因。肺实质病变主要指肺通气功能障碍,该患儿支气管镜检查未见明显气道软化等严重肺实质病变,考虑肺间质性病变可能性大。该患儿胸部X线检查主要表现为双肺呈透亮度减低、细颗粒状改变,且胸部CT呈现典型“铺路石征”(小叶间隔增厚伴有斑片状磨玻璃影)[8-9],这对PAP 的诊断具有重要的提示作用。该患儿起病年龄早,且有不良家族史,考虑先天性PAP 可能性大。先天性PAP 是一种罕见的肺部疾病,以难治性呼吸窘迫综合征为主要表现,当患儿出现常规治疗效果不佳的肺间质病变时需警惕该疾病。患儿住院期间完善支气管镜检查,可见BALF外观呈牛奶样浑浊[4],且PAS 染色阳性,具有诊断价值;结合基因检测结果回报存在ABCA3基因复合杂合突变,患儿先天性PAP诊断明确。在治疗方面:PAP的治疗目标是减轻症状、改善氧合及提高生活质量。全肺灌洗疗法(whole lung lavage, WLL)是目前PAP 的一线治疗方案。WLL 是一种侵入性操作,通过移除肺泡内的磷脂和脂蛋白来改善症状,也仅是对症治疗;但大多数先天性PAP 患者是在新生儿期起病,目前WLL 尚不适用于新生儿。药物治疗是期望通过免疫抑制疗法减少肺纤维化的发生,常用的免疫抑制药物包括糖皮质激素联合羟氯喹[7];但药物治疗在临床应用中的治疗效果及益处仍有限,且需密切注意激素及羟氯喹应用的不良反应。针对发病原因,近年来有吸入GMCSF、肺巨噬细胞移植、体外造血干细胞基因治疗等新方法被报道,但这些治疗方法多用于原发性PAP 及成人治疗,新生儿领域内尚未见报道[6,10-11]。Cho 等[12]报道1例新生儿期起病的PAP采用静脉注射免疫球蛋白治疗获得满意疗效,但该患儿的免疫检查及基因检测均未见明显异常。本例患儿年龄小,体重轻,体格条件暂时无法行WLL 治疗,建议使用免疫抑制治疗,即静脉甲泼尼龙冲击治疗后序贯口服甲泼尼龙,同时加用羟氯喹口服治疗。
3.3 影像科会诊
不同病因导致的PAP,其典型的胸部CT 表现相似,主要为双肺片状磨玻璃影与增厚的小叶间隔交替重叠,形成“铺路石”样或“烂石路”样改变[8]。其形成主要是由于肺泡腔和终末支气管内完全或者部分填充大量富含磷脂和蛋白质的颗粒状物质,导致肺泡密度增高,这些异常改变和正常肺组织同时存在导致影像学表现为实变或磨玻璃影;同时病灶部位的肺组织间隔产生轻微炎症反应,导致小叶间隔增厚[13]。病灶内蛋白样物质沉积较多或伴有感染时,磨玻璃影密度增高,局部呈肺实变,其中可见支气管充气征[14-16]。该患儿胸部CT 表现为双肺透亮度明显减低,见弥漫斑片状、片状磨玻璃密度影及网格影,局部见支气管充气征,符合典型PAP表现。
3.4 病理科会诊
肺组织病理学结果可通过肺活检获得,大龄儿童可经支气管镜行支气管活检取得,但临床上一般可通过基因检测、影像学检查及BALF检查诊断PAP,不需要肺组织病理学检查。BALF 是PAP细胞病理学诊断最重要的样本[17]。PAP患者BALF肉眼观为乳白色,静置或离心后可获得深褐色沉淀物,光镜观察可见均质嗜伊红性细胞颗粒状脂蛋白性物质,有时可见针状裂隙,免疫组化染色提示PAS 染色阳性[18]。该患儿BALF 外观为乳白色,细胞特殊染色提示PAS 阳性,符合PAP 疾病的病理特点。
3.5 分子诊断中心会诊
ABCA3基因定位于16 号常染色体上,是参与内源性脂质跨膜转运的ABCA转运家族的一员,能够编码合成1 704个氨基酸的ABCA3蛋白。ABCA3蛋白是膜整合蛋白,它利用水解ATP 的能量对溶质中各种生物分子进行跨膜转运,其主要在肺泡Ⅱ型细胞中表达,并定位于板层小体的膜上,板层小体是溶酶体衍生的细胞内储存细胞器,肺表面活性物质与ABCA3 蛋白合成包装后进入板层小体并经胞吐作用排到肺泡表面[19]。目前已经发现有150 余种关于ABCA3基因的突变,包括无义、移码、错义、剪接位点和插入/缺失等[20]。由ABCA3突变引起的肺部疾病以常染色体隐性方式遗传,需要两个等位基因都发生突变。该患儿母亲携带该致病基因的点突变,父亲携带该基因内片段缺失,符合遗传模式;且该患儿有一哥哥生后因呼吸衰竭不久即夭折,推测其可能患该类疾病。
3.6 胎儿医学科和产前诊断中心会诊
ABCA3基因突变引起的PAP 预后根据突变类型不同而有不同。有些在婴儿期或新生儿期即发生严重的不可逆呼吸衰竭而死亡,也有患者表现为慢性静止性或进行性间质性肺病,研究发现约50%的患者能够在未进行肺移植的情况下存活到第2 个10 年[21]。本例患儿具有不良家族史,患儿有一哥哥生后不久即因呼吸衰竭夭折,应尽早对先证者进行病因及基因筛查,以便发现致病变异。ABCA3基因突变引起的肺部疾病的遗传方式为常染色体隐性遗传。本例患儿的母亲携带该基因点突变,父亲携带基因内片段缺失,患儿为复合杂合突变,符合遗传模式。若父母双方为常染色体隐性遗传性疾病携带者,子女患病风险为25%,表型正常的子女有2/3的风险为携带者[22]。随着产前基因诊断方法的不断发展,ABCA3基因突变引起的先天性PAP 进行产前基因诊断已成为可能。自然受孕后,在孕早期采集绒毛组织,孕中期采集羊水细胞或脐血进行产前基因诊断;辅助生殖可在胚胎植入前行遗传学产前诊断,最终达到早诊断、早干预的目的。
3.7 新生儿重症监护病房诊断思路总结
该患儿生后早期起病,以“呼吸困难、呼吸衰竭”为主要表现,入院后行胸部CT 及BALF 病理学检查,结果提示PAP。通过基因检测明确诊断为由ABCA3基因突变导致的先天性PAP。既往临床病例分析表明,约39%的PAP 患儿以呼吸困难起病[23]。先天性PAP 一般发生更早,多在新生儿期起病。先天性PAP 是足月儿常见的引起呼吸困难的疾病,常规治疗无效的呼吸困难需考虑该疾病[24-25]。当临床考虑该病可能性大时需尽早协同影像科医生进行胸部CT 扫描,并协同呼吸科医生进行支气管镜检查,同时对BALF进行病理学检测并注明需PAS 特殊染色,可早期进行临床诊断;并且在呼吸科医生协助下早期开始针对性治疗。不同基因导致的先天性PAP 临床转归及预后情况不同,对症治疗及免疫抑制治疗是目前的主要治疗方法。但该疾病总体预后不佳,尤其是新生儿期起病的患者,预后不良,治疗手段有限。
4 住院经过及转归
入院时根据病史及临床表现初步诊断为“新生儿呼吸衰竭、先天性肺发育异常、新生儿肺炎、宫外发育迟缓、肺动脉高压、卵圆孔未闭”,予积极寻找病因并对症支持治疗。入院后因高流量鼻导管吸氧状态下呼吸困难明显,予气管插管、有创呼吸支持及布地奈德雾化治疗。结合胸部CT、BALF病理学检查结果和基因检测结果,诊断为先天性PAP。在呼吸科会诊协助诊治下,予甲泼尼龙冲击治疗后序贯甲泼尼龙口服,并同时口服羟氯喹治疗原发病。随访胸部X线检查,显示肺部渗出及间质性改变较前好转,但呼吸机支持氧需求仍较高(FiO270%~80%,呼气末正压6~7 cmH2O,气道峰压21~22 cmH2O)。同时予高热卡配方奶改善营养状态,大剂量激素治疗过程中补充钙、磷、维生素D,并予奥美拉唑保护胃黏膜。
考虑该病目前尚无特效治疗方法,远期预后不佳,向患儿家属充分告知病情、转归,并进行遗传咨询后,家属选择终止治疗。患儿于终止治疗当天死亡,死亡日龄71 d。尸检报告结果提示患儿存在肺间质病变伴炎症细胞浸润,光镜观察可见均质嗜伊红性细胞颗粒状脂蛋白性物质,免疫组化染色提示PAS 染色阳性,支持先天性PAP 的诊断。
5 小结
目前国内关于先天性PAP的病例报道很稀少,主要是由于PAP 的症状缺乏特异性,导致其容易被漏诊或延误诊断。对于每一个持续呼吸窘迫、低氧血症的婴儿,在予肺表面活性物质、机械通气等对症治疗后效果不佳,且常规检查不能解释患儿的呼吸衰竭时需警惕先天性PAP 可能。如有类似病史的兄弟姐妹或近亲父母更强烈提示PAP的诊断。先天性PAP 临床表现无明显特异性,多起病于新生儿期,主要为呼吸窘迫综合征样表现,包括呼吸困难、发绀、气促等。根据基因异常的不同临床进展情况存在差异[7],SFTPB突变常导致肺表面活性蛋白-B 完全缺乏,起病更为急骤,常在生后数小时即出现需要机械通气的呼吸困难;SFTPC突变常合并发育迟缓与消化功能异常;NKX2-1突变者则表现多样,可合并甲状腺功能异常和神经系统异常综合征;ABCA3突变者的临床表现与突变方式密切相关,与杂合突变者相比,纯合突变者起病较早且临床表现常较重,与SFTPB突变表现相似。目前先天性PAP 诊断主要依赖BALF检查及基因检测,因此,一旦怀疑该疾病应尽早完善上述检查。但该病在新生儿期除对症治疗外,尚无明确有效的治疗方法。因此,诊断先天性PAP后需进行遗传咨询。
利益冲突声明:所有作者声明无利益冲突。
