细胞焦亡在糖尿病心肌病中作用与机制的研究进展
2023-10-31王敢钟江华陈小盼
王敢 钟江华 陈小盼
糖尿病是一种由于胰岛素分泌不足、胰岛素抵抗或两者共同作用,导致以血糖水平升高为主要表现的慢性代谢性疾病[1]。随着经济的发展,糖尿病的发病率不断上升,全球现约有超过5亿糖尿病患者,全球糖尿病的患病率目前估计超过10%[2]。糖尿病也成为了最常见且最严重的慢性疾病之一。长期血糖升高,还导致微血管及大血管受损,也引发了各种并发症。糖尿病心肌病(diabetic cardiomyopathy,DCM) 是指糖尿病患者在高血糖、胰岛素抵抗、氧化应激及炎症等因素共同作用后,出现的一种表现为心肌结构和功能异常的特殊心肌病变,其发生独立于冠心病及高血压等其他心血管危险因素[3]。DCM被认为是糖尿病最常见的并发症之一,增加了出现心力衰竭的风险[3]。
自DCM被提出以来,其发生机制备受研究者们关注。在DCM发生发展过程中,各种代谢紊乱可促进心脏结构改变以及心脏功能下降,主要包括胰岛素信号传导受损、脂肪酸氧化减少、线粒体功能障碍以及糖基化反应增加等[4-5]。与此同时,肾素-血管紧张素-醛固酮系统(renin-angiotensin-aldosterone system,RAAS)的激活、炎症、内皮细胞功能障碍、细胞死亡等也促进了DCM的发生发展[4-5]。但迄今为止,DCM的发病机制尚未完全明确。有研究者发现,DCM的发生过程中可能存在多种细胞死亡形式[6]。细胞死亡的调控或许是DCM的潜在治疗靶点[6]。细胞焦亡作为细胞死亡的一种类型,可能是推动DCM发生发展的重要原因之一。本文就细胞焦亡在DCM中的作用作一综述。
1 细胞焦亡
细胞焦亡是一种伴随有炎症反应的细胞程序性死亡方式。与细胞凋亡不同,细胞焦亡可引起细胞肿胀、质膜破裂形成膜孔,释放白细胞介素(interleukin,IL)-1β和IL-18等促炎因子及细胞内容物[7]。细胞焦亡可调节并放大炎症反应,在多种疾病中发挥重要作用[7]。细胞焦亡主要由Gasdermin家族介导,Gasdermin家族主要由Gasdermin A、Gasdermin B、Gasdermin C、Gasdermin D、Gasdermin E和DFNB59六种蛋白质组成[7-8]。除了DFNB59外,该家族成员均有两个保守结构域:N端的成孔结构域和C端的阻遏结构域,可诱导细胞焦亡[7-8]。
目前研究结果表明,细胞焦亡有以下几种途径:半胱氨酸天冬酶1(Caspase-1)介导的规范途径、Caspase-4/5/11介导的非规范途径、Caspase-3和Caspase-8介导的途径(图1)[7-8]。(1)在规范途径中,当宿主细胞受体识别各种刺激因素时,主要包括病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)和损伤相关分子模式(damage associated molecular patterns,DAMPs),可促进下游的pro-Caspase-1激活为成熟的Caspase-1,同时促进炎症小体的组装[7-8]。成熟的Caspase-1可裂解Gasdermin D(GSDMD)形成N-GSDMD。随之GSDMD的N端成孔结构域可非选择性地穿透细胞膜形成膜孔,进一步导致细胞肿胀、裂解、死亡[7-8]。(2)在非规范途径中,Caspase-4/5/11可通过自身的半胱氨酸天冬酶募集域蛋白(caspase recruitment domain protein,CARD)直接结合细胞内的脂多糖(lipopolysacchatide,LPS)来实现激活[7-8]。激活后的Caspase-4/5/11同样可裂解GSDMD形成N-GSDMD,从而介导细胞焦亡的发生。(3)与规范途径和非规范途径不同,人们发现了由Caspase-3介导的焦亡途径。激活后的Caspase-3通过切割GSDME,促进N-GSDME结构域到细胞膜处介导膜孔形成,从而导致细胞焦亡的发生[7-8]。(4)有研究者发现,被认为是细胞凋亡促进剂的Caspase-8也可切割GSDMD参与细胞焦亡[7,9]。虽然这四种途径各有不同之处,但是其又相互关联,它们最终均介导了膜孔的形成,导致细胞破裂,释放了大量的促炎因子,如IL-1β和IL-18[7-9]。
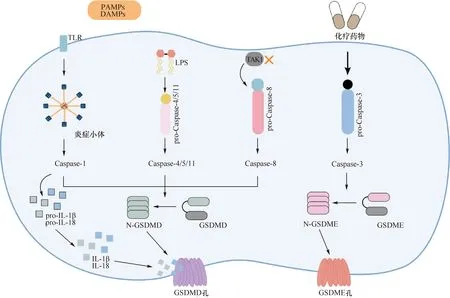
PAMPs:病原体相关分子模式;DAMPs:损伤相关分子模式;TLR:Toll样受体;Caspase:半胱氨酸天冬酶;IL:白细胞介素;LPS:脂多糖;TAK1:转化生长因子活化激酶1图1 细胞焦亡的四种不同途径
2 细胞焦亡与DCM
1型糖尿病与2型糖尿病有着不相同的发病机制。但是相关研究证实,细胞焦亡作为一种伴有炎症反应的细胞程序性死亡方式,其在两者及糖尿病并发症的发生发展中均发挥了重要作用[10-11]。研究已经证实,慢性炎症是导致DCM的重要原因之一,慢性炎症可通过触发NLRP3等炎症小体的激活,从而导致细胞发生焦亡[10-11]。NLRP3炎症小体是一种由含pyrin结构域NOD样受体蛋白3 ( NOD-like receptor thermal protein domain associated protein 3,NLRP3)、含有CARD的凋亡相关斑点样蛋白(apoptosis associated speck like protein containing a CARD,ASC)、Caspase-1构成的多聚体蛋白复合物[10-12]。有学者认为,NLRP3炎症小体的激活是细胞焦亡的关键步骤,其在细胞焦亡促进DCM等糖尿病并发症的发生发展中也发挥着必不可少的作用[10-12]。与其他心肌病不同的是,在DCM中,NLRP3炎症小体的激活机制主要依靠高血糖环境下大量活性氧(reactive oxygen species,ROS)的产生,大量ROS促使硫氧还原蛋白相互作用蛋白(thioredoxin-interacting protein,TXNIP)与NLRP3结合从而促进NLRP3炎症小体的激活,诱导细胞发生焦亡[13]。细胞焦亡可在内皮细胞、心肌细胞、平滑肌细胞等多种细胞中发生,并促进了DCM发生的病理生理过程[14]。研究细胞焦亡的不同途径和发生细胞焦亡的细胞类型对DCM的影响可以为DCM提供可能的治疗靶点。
3 与DCM相关的细胞焦亡通路
3.1 Toll样受体4(Toll-like receptor 4,TLR4)/核因子-κB(nuclear factor kappa-B,NF-κB)/NLRP3炎症小体通路

TLR4:Toll样受体4;TRIF:β干扰素TIR结构域衔接蛋白;MyD88:骨髓分化因子88;NF-κB:核因子κB;CBP:环磷酸腺苷反应元件结合蛋白(CREB)的结合蛋白;Caspase:半胱氨酸天冬酶;IL:白细胞介素;TXNIP:硫氧还原蛋白相互作用蛋白;ROS:活性氧;Nrf2:核因子红细胞2相关因子2;Keap 1:Kelch样环氧氯丙烷相关蛋白1;ARE:抗氧化反应元件;HO-1:血红素加氧酶1;GCLC:谷氨酸-半胱氨酸连接酶催化亚基图2 与糖尿病心肌病相关的细胞焦亡通路
细胞焦亡主要通过NLRP3炎症小体及活化的Caspase-1来发挥作用[12]。典型的NLRP3炎症小体激活主要包括启动和激活两个步骤。其中启动主要通过NF-κB通路来实现[15]。TLR4、骨髓分化因子88(myeloid differentiation factor88,MyD88)、NF-κB与NLRP3炎症小体的激活密切相关[15]。激活后的NLRP3炎症小体可加速成熟Caspase-1的形成,裂解生成大量的IL-1β和IL-18通过膜孔介导细胞焦亡[7-9,15]。通过高能量喂养大鼠后腹腔内注射链脲佐菌素和使用高葡萄糖诱导H9C2心肌细胞分别制作DCM的体内体外模型,发现DCM模型组TLR4、MyD88和NF-κB p65 的mRNA表达均较对照组显著增加[16]。此外,DCM大鼠血清中的肿瘤坏死因子(tumor necrosis factor,TNF)-α、IL-1β和IL-6水平也较正常喂养的大鼠组升高[16]。同时还有研究者发现,TLR4-NF-κB通路下游的NLRP3炎症小体在DCM组中也显著增加[17]。另外,使用药物抑制该通路可减少细胞焦亡,减弱DCM大鼠的心脏炎症,改善心脏功能及心脏不良重构[16-18]。以上实验均表明,TLR4/NF-κB/NLRP3炎症小体通路在DCM的发生发展过程中具有推动作用,抑制该通路的某一环节或许可以为DCM提供新的治疗手段(图2)。
3.2 ROS/TXNIP/NLRP3炎症小体通路
ROS是线粒体呼吸或代谢过程中由特定酶产生的副产物[19]。ROS具有两面性,其具有有益作用的同时也会对机体造成损害[19]。长期的高血糖、高血脂和慢性炎症等不良环境,会导致产生ROS的酶活性上调,产生大量的ROS,诱发氧化应激[19-20]。在糖尿病患者和糖尿病大鼠模型中,研究也均可发现其ROS水平显著上升[21-22],而大量ROS的生成及氧化应激会导致心脏不良重构,加速DCM的进程[18-19]。TXNIP是葡萄糖代谢及脂质代谢的主要调节因子之一[23]。TXNIP已被证明是依赖ROS的NLRP3炎症小体激活调节因子,大量的ROS可促进TXNIP与NLRP3的结合,从而触发NLRP3炎症小体的激活[23-24]。在大鼠INS-1胰岛β细胞相关研究中,高血糖显著增加了细胞中TXNIP、NLRP3和Caspase-1的表达,且TXNIP具有葡萄糖剂量依赖性,随着葡萄糖浓度的增高,其表达也增加[25]。这也表明糖尿病也可能通过ROS/TXNIP/NLRP3炎症小体通路触发细胞焦亡,从而加快DCM进程。
3.3 其他与DCM相关的焦亡途径
研究者们还发现了其他与DCM相关的焦亡途径。核因子红细胞2相关因子2(nuclear factor erythroid-2-related factor 2,Nrf2) 是一种转录因子,在多种组织和器官中均有表达。研究结果证实,Nrf2信号通路与各种心脏疾病密切相关,其可通过控制各种抗氧化基因发挥抑制氧化应激的作用来调节心脏稳态[26]。Nrf2与其阻遏因子Kelch样环氧氯丙烷相关蛋白1(Keap1)结合并存在于细胞质中[26-27],但大量ROS和氧化应激会使Nrf2与Keap1解离,使Nrf2转移至细胞核中与抗氧化反应元件(antioxidant response element,ARE)的启动子区域结合,激活抗氧化基因,促进抗氧化酶的产生。这些抗氧化酶具有抑制炎症及抗氧化作用,保护心肌细胞防御糖尿病和高糖氧化带来的心肌损伤[27]。Nrf2激活后还可上调其下游抗氧化因子血红素加氧酶1(heme oxygenase 1,HO-1)的表达水平,清除细胞内堆积的ROS[27]。除此之外,Nrf2也是NF-κB的上游调控因子之一[24,28-29]。激活后的Nrf2可抑制NF-κB从而减少NLRP3炎症小体的生成,抑制细胞焦亡,从而保护糖尿病大鼠模型的心肌细胞[28]。因此,Nrf2/HO-1/NF-κB/NLRP3炎症小体通路也可能在DCM的细胞焦亡过程中也起着重要作用。
3.4 发生焦亡的细胞类型对DCM的作用
细胞焦亡可发生在心脏的各种细胞中,包括心肌细胞、巨噬细胞、成纤维细胞及内皮细胞等。在高血糖环境下,促使心脏各类细胞发生细胞焦亡,这也加快了DCM的发展进程[13]。
心肌细胞的死亡,无论其是急性的还是慢性的,均在DCM的进程中起重要推动作用[30]。新的证据也证实,由NLRP3炎症小体诱导的心肌细胞焦亡也是DCM进展中的关键步骤[13,30]。糖尿病小鼠模型较非糖尿病小鼠来说,其心肌细胞中NLRP3炎症小体、Caspase-1显著上升,并伴有明显的细胞焦亡[31]。在压力超负荷诱导心肌细胞焦亡的实验中,还发现细胞焦亡可促进心肌细胞发生肥大,导致心脏不良重构。另外,通过抑制细胞焦亡,可发现由压力超负荷引起的心脏肥大和心功能不全得到改善[32]。
巨噬细胞焦亡也在由高血糖引起的并发症中发挥了重要作用[33]。巨噬细胞是免疫系统的重要组成成分,它具有识别、吞噬、分泌作用,可调节免疫系统并维持体内平衡[34-35]。一项研究发现在糖尿病小鼠模型中,持续的高血糖状态可导致巨噬细胞功能障碍,而NLRP3炎症小体的活化在其中起着重要作用。通过抑制组织蛋白酶B和NLRP3炎症小体的活化可恢复由高血糖诱导的部分巨噬细胞功能障碍[33]。同时还有研究者发现,在卒中后的糖尿病小鼠模型中,NLRP3炎症小体在M1巨噬细胞中激活后可加重心功能障碍,促进心肌发生形态学改变,且使用CY-09抑制NLRP3炎症小体后可改善糖尿病小鼠的心脏功能[35]。以上研究表明,巨噬细胞中NLRP3炎症小体的激活和细胞焦亡可能在DCM的发生过程中发挥着重要作用。
部分DCM除了可表现为心肌细胞肥大外,还可表现为心脏间质纤维化[36]。心脏成纤维细胞是心脏的重要组成,是心肌纤维化过程中的效应细胞[36]。在糖尿病状态下,糖基化终末产物(advanced glycation end products,AGEs)等代谢产物可促进成纤维细胞分化成肌成纤维细胞[5,36]。在心脏的纤维化过程中,细胞焦亡也发挥了重要作用。糖尿病大鼠的心脏组织中Caspase-1、GSDMD、胶原蛋白Ⅰ和胶原蛋白Ⅲ的mRNA水平较正常小鼠明显上升,而使用药物抑制糖尿病大鼠的焦亡可阻碍心脏纤维化的发生[37]。这也表明心脏成纤维细胞焦亡在DCM的发生发展中也起着重要的驱动作用。
除此之外,血管内皮细胞也在DCM的发生发展过程中发挥了重大作用。血管内皮细胞功能障碍也是DCM发生发展的关键病理基础[38]。血管内皮细胞在循环血液和血管外基质之间形成了一层半透性屏障,该内皮屏障可调节细胞连接来维持机体稳态[39]。在DCM发展过程中,活化的NLRP3炎症小体诱导细胞发生焦亡释放大量促炎因子IL-1β和IL-18。这些促炎因子随之与细胞表面受体相结合,增强了内皮中粘附因子和趋化因子的表达,同时增加白细胞的粘附和外渗。最终导致细胞间连接破坏、内皮屏障功能障碍、血管通透性增加,这也有利于促炎细胞和促炎因子渗透,加速心脏的不良重构[30,38-39]。另外,还有研究者发现,与NLRP3相关的细胞焦亡不仅损害心脏内皮细胞,而且可降低心脏的微血管密度,而这种效应可通过抑制NLRP3炎症小体及其相关的细胞焦亡来抵消[40]。
4 DCM中细胞焦亡的调控
目前,越来越多的证据表明细胞焦亡在DCM的发生发展过程中起重要推动作用。调节DCM的细胞焦亡和焦亡相关通路或许可以为DCM的防治提供新的治疗方向。同时随着研究的不断进展,目前发现一些药物及物质可通过靶向NLRP3炎症小体/焦亡来治疗DCM。
4.1 降糖药物
高血糖是NLRP3炎症小体的激活并诱导细胞焦亡的关键因素。多种降糖药物也具有抑制细胞焦亡的作用。二甲双胍是一种使用较为广泛的降糖药物,可激活AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)下调NLRP3、Caspase-1和IL-1β的表达,从而改善糖尿病小鼠的心脏功能[41]。钠-葡萄糖共转运蛋白2抑制剂(sodium-dependent glucose transporters 2 inhibitor,SGLT2i)是一种新型降糖药物,其通过抑制肾近端肾小管葡萄糖重吸收和增加尿糖排泄来发挥降糖作用。恩格列净是SGLT2i药物的一种,其对糖尿病人群具有心血管益处。研究证实,恩格列净可通过可溶性鸟苷酸环化酶(soluble guanylate cyclase,sGC)-环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)-cGMP依赖的蛋白激酶(cGMP-dependent protein kinase,PKG)途径保护心脏功能,延缓DCM的进展[42]。除此之外,在非糖尿病模型中,恩格列净也可通过参与NLRP3和MyD88相关途径缓解心脏炎症、减弱心肌纤维化[43]。
4.2 NLRP3炎症小体抑制剂
NLRP3炎症小体是焦亡发生的重要中间一环。抑制NLRP3炎症小体可有效调控细胞焦亡改善DCM。MCC950是一种小分子NLRP3炎症小体抑制剂,可通过阻断ASC寡聚化、抑制NLRP3炎症小体活化。其被证实可改善糖尿病血管内皮功能障碍[44]。除此之外,在糖尿病性脑病中,MCC950也可通过抑制NLRP3炎症小体,降低IL-1β等改善糖尿病性脑病[45]。
4.3 非编码RNA(non-coding RNA,ncRNA)可调节DCM的细胞焦亡
在细胞焦亡加速DCM发生发展的过程中,ncRNA也起了重要调节作用。ncRNA包括很多类型,其主要有microRNA(miRNA)、长链非编码RNA(long ncRNA,lncRNA)和环状RNA(circular RNA,circRNA)[46]。miRNA是一类长度约为20个核苷酸的内源性小非编码RNA。在体内及体外实验中证实,糖尿病组的miRNA-30d表达显著增加。其表达增加上调了Caspase-1和促炎因子的表达[47]。另外,lncRNA也可介导细胞焦亡来影响DCM的进程。相关实验证实,lncRNA KCNQ1OT1在糖尿病患者、高糖诱导的心肌细胞和糖尿病小鼠模型中表达增加。沉默KCNQ1OT1可通过靶向miR-214-3p和Caspase-1来抑制细胞焦亡,同时还可以改善细胞骨架结构的异常和钙超载,改善心脏结构和功能[48]。circRNA是各种疾病的关键调节因子。circRNA DICAR被证实可以缓解DCM,而DICAR的敲除可增强DCM中的细胞焦亡[49]。
5 展望
综上所述,细胞焦亡作为一种伴有炎症的细胞死亡方式,其在DCM的发生发展过程中具有重要作用。了解与DCM相关的不同细胞焦亡途径及不同细胞焦亡对DCM的影响有助于我们寻找新的治疗靶点。目前也有研究者发现,通过降血糖药物和靶向NLRP3炎症小体的抑制剂等干预细胞焦亡可延缓DCM的进展[41-43]。但DCM与细胞焦亡之间关联的确切机制暂也尚未明确,仍需广大研究者进一步探索。
利益冲突:无
