流动注射在线分析法测定水源水中的总氮
2023-10-25贺舒文褚莹倩崔晗曹文军
贺舒文,褚莹倩,崔晗,曹文军
(大连海关技术中心,辽宁大连 116400)
水中总氮包括硝酸盐氮、亚硝酸盐氮、溶解态氨、无机铵盐及有机含氮化合物中的氮,水中总氮超标,易造成浮游植物过度繁殖,水体富营养化[1],严重时能够引发赤潮[2-5],因此总氮是评价水体污染[6]和自净状态的重要指标之一[7-8],总氮的监测已成为水体保护的重要研究课题[9]。由于目前总氮检测设备存在体积大、检出限高、检测时间长和废液排放量大等缺陷[10-11],对于低检出限、自动化程度高、废液排量少和检测效率高的总氮分析方法的开发和优化具有很重要的现实意义。
总氮的分析方法主要有紫外可见分光光度法[9]、离子色谱法[12]、电极法[13]、气相分子吸收光谱法[9]、流动注射法[8]等。分光光度法操作步骤繁琐,耗时长,重复性差,而且不能满足批量样品的快速测定;离子色谱法和电极法的仪器操作简单,且能够进行连续测定,但样品处理步骤较多,检测灵敏度较低,检测成本较高,检测大批量样品费时费力;气相分子吸收光谱法需要用氮气作为载气进行检测,而且试剂用量大[14],相对增加了操作步骤和检测成本。近年来流动注射在线分析技术被广泛地应用于水质分析中,并得到推广[15-19]。流动注射在线分析法是一种自动化程度高、分析速度快、准确度和精密度高的分析技术[20];该方法减少了人为操作带来的实验误差和环境污染,实现了实验室检测的自动化,提高了工作效率。
目前,系统的针对水源水中总氮检测样品处理中的水样浊度干扰、实验用水、试剂纯度选择、试剂配制及存放、气泡的干扰、干扰物的消除、显色剂浓度优化等对实验结果影响的研究较少,同时做到高回收率和精密度等结果的测定方法尚未见报道。笔者结合相关标准和研究成果,有针对性的对样品处理的相关步骤进行探索和优化,建立了流动注射在线分析法测定水源水中总氮的方法,测定结果满足相关技术要求,该方法简便快速,自动化程度高,试剂消耗少,精密度和准确度高,并且能够满足大批量水源水样品的分析需求。
1 方法原理
样品中的总氮于90 ℃高温下被过硫酸盐和紫外灯(UV-254 紫外灯)在线消解为硝酸根,再通过镉柱还原为亚硝酸根,亚硝酸根与磺胺产生重氮化反应,生成重氮离子,重氮离子与萘乙二胺盐酸盐结合产生一种紫色物质,在波长540 nm 处测定,根据朗伯-比尔定律,经仪器处理后得到样品峰面积,由标准曲线法计算目标物中的总氮含量。
2 实验部分
2.1 主要仪器与试剂
流动注射分析仪:QC8500 型,带UV-254 紫外灯的样品预处理模块,美国哈希公司。
超声波水浴清洗器:KQ-700DV 型,频率范围为0~100 kHz,上海一恒科学仪器有限公司。
pH计:测量范围为0.00~14.00,梅特勒-托利多仪器(上海)有限公司。
总氮标准溶液:质量浓度为1 000 mg/L,标准物质编号为GSB04-2837-2011 (b),北京有色金属研究总院。
氯化铵、磺胺、盐酸萘乙二胺、磷酸、氢氧化钠、过硫酸钾、四硼酸钠、焦亚硫酸钠、硫酸、乙二胺四乙酸二钠:优级纯,国药集团化学试剂有限公司。
水性滤膜:孔径为0.45 μm,美国哈希公司。实验用水为GB/T 6682—2008 规定的一级水。
2.2 标准溶液配制
总氮标准工作溶液:用实验用水作溶剂,采用逐级稀释的方式将总氮标准溶液配制成质量浓度分别为0.000、0.050、0.200、1.00、2.00、5.00、10.0 mg/L 的氮系列标准工作溶液。
2.3 仪器工作条件
检测光程:10 mm;检测波长:540 nm;进样体积:68 μL;蠕动泵转速:35 r/min;超声除气时间:30 min;超声频率:40 kHz。
2.4 检测流程
流动注射在线分析法测定水源中总氮流程图如图1所示。
2.5 样品处理
样品检测前需用0.45 μm的水性滤膜过滤。如需将水样保存24 h以上,则必须添加固定剂硫酸调节pH为1~2后冷藏保存于硬质玻璃瓶中,并在7日内完成检测[21],或者保存在聚乙烯瓶中,于-20 ℃冷冻,可保存1个月[22]。
添加固定剂保存的样品在检测前应该调节pH,即取100 g/L 氢氧化钠溶液约0.55 mL 于100 mL 水样中,使样品pH保持在7左右。
2.6 标准曲线绘制
由自动进样器依次从低浓度到高浓度取样,测定总氮系列标准工作溶液,以总氮质量浓度(mg/L)为自变量、峰面积为因变量进行线性回归,得标准曲线方程。
2.7 水样测定
按照与制作校准曲线相同的实验条件进行水样测定,同时做空白试验,以标准工作曲线外标法进行定量。
2.8 分析结果的表述
2.8.1 结果计算
水源水中总氮的含量 (以氮的质量浓度ρ计,mg/L)按照式(1)计算:
式中:y——信号值(峰面积);
a——线性方程的截距;
b——线性方程的斜率;
f——样品溶液稀释倍数。
2.8.2 结果表示
当总氮的测定值小于1.00 mg/L时,测定值保留到小数点后第3位;当测定值大于或等于1.00 mg/L时,测定值保留到小数点后第2位。
3 结果与讨论
3.1 水样浊度的干扰
相关研究表明,水样的浊度去除效率由高到低依次为离心、0.45 μm滤膜、定量滤纸[23],而且三种处理方式都能保证检测结果的准确性。在实际检测工作中,为了简化操作步骤和节约成本,选用0.45 μm的水性滤膜来去除水源水中浊度的干扰,该方法操作步骤简单、所需时间较少、干扰较小,而且适用于大批量水源水样品的分析。
3.2 实验用水的选择
水中总氮检测的标准方法《水质 总氮的测定碱性过硫酸钾消解紫外分光光度法》(HJ 636—2012)中规定实验用水须为无氨水或新制备的去离子水,考虑到实际工作的可操作性,以及无氨水在制备过程中容易被蒸馏装置污染,且效率较低,在实际检测中使用较少,所以选用去离子水作为实验用水,去离子水的纯度比较高,且随用随制,不易受到氨的污染,能够在保证检测结果准确性的同时兼顾工作效率。
3.3 试剂纯度选择
在水源水中总氮的检测中,试剂的纯度会影响检测结果的准确性,其中过硫酸钾试剂的纯度是直接影响总氮测定结果准确度的关键因素。由于分析纯试剂的空白值明显高于优级纯试剂的空白值[24],所以在进行水源水中总氮的检测时应选用优级纯试剂,而且应该对新采购的试剂进行空白试验。
3.4 试剂器皿的洗涤
水源水中总氮检测用的容量瓶、烧杯等玻璃器皿的洁净程度对检测结果具有一定的影响,如果仅用水冲洗玻璃器皿的话,其空白值会相对较高,不能满足标准方法对于空白值的要求,而采用盐酸溶液(体积比为1∶9)浸泡24 h后用自来水冲洗,然后用去离子水冲洗干净后立即使用,其空白值符合标准要求[25]。
3.5 试剂配制及存放
在配制试剂时,载液、缓冲溶液和显色剂等溶液按照常规步骤配制即可,而过硫酸钾和硼酸盐溶液的配制则需要注意温度的控制,因为在常温下过硫酸钾的溶解度比较低,溶解非常慢,所以在配制过程中可以将其置于水浴中加热溶解,温度一定要控制在60 ℃以内[26],因为过硫酸钾容易在60 ℃以上发生氧化分解反应,导致其氧化能力慢慢降低,从而影响检测结果。
对于试剂存放的条件和时间,《水质 总氮的测定 连续流动-盐酸萘乙二胺 分光光度法》(HJ 667—2013)中给出了相关的参考信息,但是考虑到本方法使用的载液是去离子水,应该现用现取,而缓冲溶液在使用前需要检查其pH值,显色剂需要通过观察其颜色变化来确定是否可用,因此,为了保证检测结果的准确性,建议显色剂、缓冲溶液应该现用现配。另外,相关研究和标准方法对于过硫酸钾溶液的存放时间有不同的观点[22,27],针对过硫酸钾和硼酸盐溶液的存放时间进行了试验,当过硫酸钾和硼酸盐溶液存放于25 ℃、样品中总氮的含量为0.10 mg/L 时,考察不同存放时间对总氮测定值的影响,试验结果见表1。
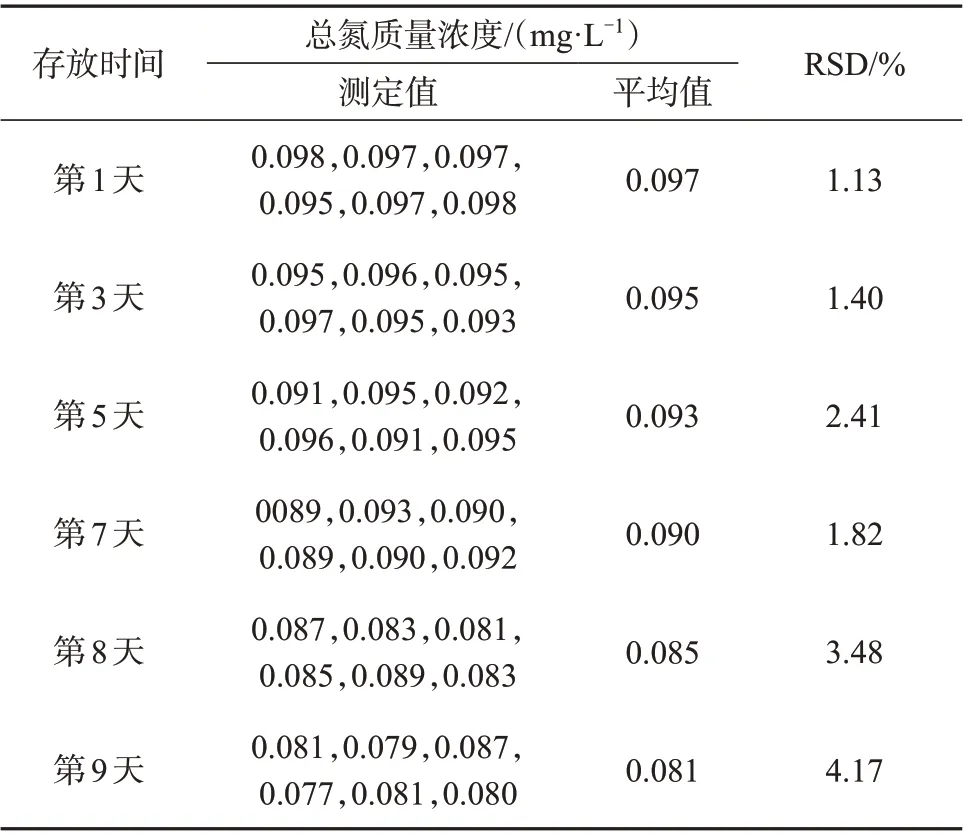
表1 过硫酸钾和硼酸盐溶液不同存放时间下总氮测定结果
从表1 测定结果来看,总氮测定值随着存放时间的延长而逐渐变小,且测定结果在第1天到第7天时波动较小,从第8 天开始波动较大,因此,为了保证检测结果的稳定性和准确性,过硫酸钾和硼酸盐溶液的存放时间应该控制在7天之内。
3.6 气泡的干扰
试剂配制过程中通常会有气泡存在,这会导致分析谱图基线不平稳,并出现杂峰,所以配制好的试剂溶液在使用前要进行脱气处理,以免造成信号波动,影响检测结果。试验对比了氦气脱气、超声波脱气和超声波结合氦气脱气三种脱气方法,以开机预热30 min后监测仪器基线状态30 min,同时进行10次纯水空白检测结果为判定依据,研究不同脱气方法的效果。结果表明:氦气脱气在 35 min后效果良好,基线平稳无气泡峰;超声波脱气在30 min 后效果良好,基线平稳无气泡峰;超声波结合氦气脱气在20 min 后效果良好,基线平稳无气泡峰。考虑到检测时间、成本等因素,选择超声波脱气30 min 为试剂溶液脱气方法。
3.7 干扰物的消除
水样中含有氯化银和硫代硫酸盐时会降低镉柱的功能,导致检测结果偏低,因此可以通过对质控样品的检测来检验镉柱的有效性。
当水源水中3价铁离子、6价铬离子和氯离子的质量浓度分别大于180、50、5 000 mg/L时,会对检测结果产生负干扰;水源水中含有高浓度的有机物或其它还原性物质时,会与过硫酸钾发生反应,导致总氮的测定结果偏低;此时,可以通过稀释水样来减少以上干扰,同时做加标回收来确认检测结果的准确性[22]。
3.8 显色剂浓度的优化
当样品中总氮含量为0.10 mg/L,考察不同浓度显色剂磺胺和盐酸萘乙二胺对总氮测定值的影响,试验结果见表2。由表2可知,当磺胺和盐酸萘乙二胺溶液的质量浓度分别为35、1 g/L 时,总氮测定值最大,因此选用此浓度组合进行检测。

表2 不同浓度磺胺和盐酸萘乙二胺溶液时水源水中总氮测定值
3.9 镉柱活化
正常情况下镉柱的颜色为深灰色或暗黑色,镉柱的还原能力下降时,其颜色往往都会变白、变浅。镉柱颜色变浅主要是因为反应管路中存在着氧化性的物质,这些氧化性物质随流路进入镉柱中会缓慢氧化镉粒表层,进而阻挡硝酸盐与镉粒有效接触,阻断还原反应。这些氧化性物质来自于水样和试剂中,或者镉柱两端密封性不好导致空气进入或在管路排气泡过程中气泡没排净。此外,镉柱中的还原反应环境为弱碱性,在碱性溶液中,硝酸盐还原产生的Cd2+与OH-反应生成的Cd(OH)2沉淀也会阻断还原反应的进行,所以镉柱需要定期活化,以保证检测的准确性。
镉柱活化:首先用玻璃棒将镉粒与足量的4 mol/L的盐酸溶液搅拌混合,充分搅拌1 min(清除镉粒表面氧化层)后用去离子水将盐酸溶液冲洗干净,直至清洗镉粒的去离子水的pH 呈中性。然后在镉粒中加入足量的2%的硫酸铜溶液,用玻璃棒充分搅拌(向镉粒表面镀铜促进还原反应)至镉柱的颜色变为深灰色或暗黑色,再用去离子水冲去镉粒之间的红色絮状被还原的铜,最后用滤纸干燥镉粒。镉粒具有毒性,操作时应避免与眼睛和皮肤等部位接触。
镉柱的填充:将镉粒装入干燥的镉柱内,填充镉柱时应不停振摇镉柱两端以使镉粒间紧密接触,镉粒填充好后封好镉柱两端,再用注射器将氯化铵缓冲溶液注入镉柱,同时保证镉柱内不得有气泡,最后将镉柱装入分析模块,备用。
3.10 实验室环境
在进行水源水中总氮的检测时,由于检测对象是总氮,所以在试验期间,相同区域内应该禁止开展涉及氨水、硝酸和其它氨盐类的检测项目,否则会造成检测结果偏高。
3.11 线性关系、检出限和定量限
按照2.6 方法测定0.040~10.0 mg/L 系列标准工作溶液线性方程为y=0.380x2+245x-237,相关系数为0.999 9。
根据GB/T 32465—2015《化学分析方法验证确认和内部质量控制要求》中关于分析方法检出限的要求进行验证[28]。按照分析步骤,对检出限的2~5倍样品进行重复测定7次以上,对n次平行测定结果计算标准偏差S,按照LD=t(n-1,0.99)×S计算方法检出限[29],以t(7,0.99)=2.998,计算8 次平行测定的标准偏差。根据HJ 168—2010《环境监测分析方法标准制修订技术导则》的要求以4 倍检出限作为方法的定量限,试验数据见表3。

表3 水源水中总氮测定方法检出限数据mg/L
3.12 方法精密度与准确度
本方法采用水源水样品进行检测,并以1 倍定量限、10 倍定量限、与实际样品同水平浓度及其10倍浓度进行加标回收试验,按全程序每个样品平行测定6 次,分别计算平均值、相对标准偏差和回收率, 试验结果见表4。由表4 数据可知,本方法加标回收率为92.5%~105%,测定值的相对标准偏差为0.22%~1.10%(n=6),测量精密度和准确度满足相关标准的要求[22,30]。

表4 精密度和准确度试验结果
4 结语
建立流动注射在线分析法测定水源水中总氮的含量。该方法灵敏度和准确度高,稳定性好,重现性好,操作简便,试剂消耗少,自动化程度高,分析周期短,极大地节约了检测时间、节省了人力物力,能够满足水源水监测需求,对提高水源水质监测预警工作具有重要意义。
