高效液相色谱法测定化妆品中艾地苯醌
2023-10-25张小媚陈桂琴肖树雄
张小媚,陈桂琴,肖树雄
(广东省药品检验所,国家药品监督管理局化妆品风险评估重点实验室,广州 510663)
艾地苯醌,又名艾地苯,是用于治疗老年性痴呆、脑功能代谢及精神症状的药物。近年来随着对艾地苯醌研究的深入,发现其具有清除自由基、抑制脂质过氧化、抑制炎症、抑制DNA损伤、抑制黑色素生成等多种抗衰老功效,因而逐渐被运用到化妆品领域中[1]。近年来,除了大麻提取物、烟酰胺、多肽类、果酸、积雪草提取物[2]等热门功效成分,艾地苯醌也凭着优秀的抗氧化作用,成为护肤产品中很火的功效成分。
近几年,化妆品功效越来越受到消费者的重视。功效是化妆品的关键属性,也是企业创新的重要载体[3]。然而,功效原料[4]价格昂贵,有些商家为了获得利润,会进行概念性添加、虚假夸大宣传效果,欺骗消费者,但添加起效浓度以下的成分根本起不到宣称的功效作用。这种概念性添加及虚假宣称的行为严重扰乱了化妆品市场的秩序,损害了消费者的合法权益。而我国现行《化妆品安全技术规范》[5]中缺少功效成分的检测项目和方法。因此,化妆品监管急需解决的问题之一便是开发和研究功效成分的检测方法。目前,功效成分的检测方法主要有高效液相色谱法[6-8]、超高效液相色谱法[9]、质谱法[10-11]等。高效液相色谱法是检测药物中艾地苯醌的主要方法,检测化妆品中艾地苯醌的方法尚未建立[12-14]。艾地苯醌作为近年十分热门的功效原料之一,急需建立其相应的检测方法以满足化妆品监管的需要。此外,化妆品成分复杂,目标物可能存在基质干扰,因此需要优化前处理方法以排除基质干扰。
笔者建立高效液相色谱法测定化妆品中艾地苯醌的含量,并与其产品标签的成分、含量进行比对,对产品是否含有宣称功效成分、使用量是否与标签标识相符等基本情况进行考察评估,不仅为政府监管部门打击功效化妆品的虚假宣传、概念性添加的违法行为提供技术支持,而且可以此为示范,逐渐形成打击概念性添加和虚假宣称等违法行为的新的监管模式。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Shimadzu LC-20AT 型,配二极管阵列检测器(DAD),日本岛津公司。
电子分析天平:XS205DU 型,感量为0.01 mg,瑞士梅特勒-托利多公司。
超声波清洗器:M8800H-C 型,美国必能信公司。
超纯水发生器:Milli Q 2882 型,美国密理博公司。
涡旋振荡器:MS 3 digital型,德国艾卡公司。
艾地苯醌标准品:纯度(质量分数)为98.48%,上海阿拉丁生化科技股份有限公司。
甲醇:色谱纯,霍尼韦尔(中国)有限公司。
氯化钠:分析纯,广州化学试剂厂。
饱和氯化钠溶液:称取20 g氯化钠,置烧杯中,加入40 mL水,边加边振摇,静置。
化妆品样品:包括精华、面霜,市售,均标识含有艾地苯醌成分。
实验用水为Milli-Q 系统(美国密理博公司)自制超纯水。
1.2 标准溶液配制
艾地苯醌标准储备液:1 000 mg/L,准确称取艾地苯醌标准品20.0 mg(精确至 0.01 mg)于20 mL棕色容量瓶中,以甲醇溶解并定容。
艾地苯醌系列标准工作溶液:分别精密移取不同体积的标准储备液,以甲醇稀释,质量浓度分别为1、10、20、50、100、200 mg/L。
1.3 样品处理
称取化妆品样品 0.5 g(精确至0.000 1 g)于10 mL具塞比色管中,加入饱和氯化钠溶液1 mL,涡旋30 s,加入甲醇5 mL,涡旋30 s,超声提取15 min,并用甲醇定容,摇匀,静置后取上清液经0.22 μm滤膜过滤,滤液作为样品溶液。
1.4 液相色谱条件
色谱柱:OSAKA SODA CAPCELL PAK MGⅡC18柱(250 mm×4.6 mm,5 µm,日本资生堂公司);柱温:40 ℃;流量:1.0 mL/min;检测波长:278 nm;流动相:甲醇-水(80∶20);进样体积:10 μL。
1.5 样品测定
分别精密吸取艾地苯醌系列标准工作溶液和样品溶液各10 µL,经高效液相色谱仪测定,记录色谱图及光谱图,以保留时间和光谱图定性,色谱峰面积定量,绘制标准工作曲线,以外标法计算艾地苯醌的含量。
2 结果与讨论
2.1 提取条件的选择
由于艾地苯醌极难溶于水,极易溶于氯仿、甲醇,但氯仿易挥发、毒性大,因此选择甲醇作为提取溶剂。考察了甲醇、80%(体积分数)甲醇水溶液、饱和氯化钠-甲醇溶液3 种溶液体系对目标物质的提取效果。结果表明,采用甲醇或80%甲醇水溶液处理的样品基质不能较好地沉淀,且用0.22 μm 滤膜较难过滤;若样品处理时先加入1 mL饱和氯化钠溶液对样品进行预分散,再用甲醇提取,提取液经0.22 μm滤膜更容易过滤。此外,同时考察了甲醇、饱和氯化钠-甲醇作为提取溶剂时的目标物回收率,分别向膏霜、乳液、面膜、凝胶、液体水基5种空白基质中加入艾地苯醌标准储备液,配成质量浓度为50 mg/L的加标溶液,经液相色谱仪测定,计算回收率,结果见表1。由表1可知,两种提取溶剂在不同基质中的回收率均大于100%,综合考虑样品处理过程的便利性,最终选取饱和氯化钠-甲醇的溶液体系作为提取试剂。

表1 不同提取溶剂艾地苯醌的回收率%
此外,考察了超声时间对测定结果的影响。选取含有艾地苯醌的膏霜类阳性样品,按1.3方法用饱和氯化钠-甲醇溶液处理后,分别超声提取15、30 min 后测定并计算艾地苯醌的提取率。结果表明,样品超声15 min 和30 min 的提取效果(测得含量)没有明显差异,因此超声时间选择15 min。
2.2 色谱柱及柱温选择
分别考察Phenomenon Titank C18色谱柱(250 mm×4.6 mm,5 µm,广州菲罗门科学仪器有限公司)和OSAKA SODA CAPCELL PAK MGⅡ C18色谱柱(250 mm×4.6 mm,5 µm,日本资生堂公司)对目标物的分离效果。在其它色谱条件一致的情况下,两个品牌的色谱柱系统适用性参数(理论塔板数、对称因子、分离度等)均能满足要求,艾地苯醌的色谱峰形及峰面积均无显著差异,且目标物的色谱峰均没有基质杂质峰干扰。从检验效率及成本方面考虑,最终选取保留时间相对较短的OSAKA SODA CAPCELL PAK MGⅡC18色谱柱进行分析。
柱温是液相色谱仪的重要参数,控制柱温可提高保留时间的重现性。一般情况下,较高柱温能增加样品在流动相中的溶解度,缩短分析时间。分别考察了OSAKA SODA CAPCELL PAK MGⅡ C18色谱柱柱温为30 、35、40 ℃时对目标物分离效果的影响。结果显示,当柱温为30 、35、40 ℃时,艾地苯醌的保留时间分别为9.1、8.7、8.1 min,保留时间差别不大,且与样品基质均能较好的分离,以较短保留时间为原则,最终柱温选择为40 ℃。
2.3 流动相的选择
考察了常用流动相甲醇-水(72∶28)、甲醇-水(80∶20)和乙腈-水(72∶28)三种流动相体系对艾地苯醌色谱行为的影响。结果显示,目标物质在三种流动相系统都具有良好的保留效果。但以甲醇-水(80∶20)或乙腈-水(72∶28)作流动相时目标物质的保留时间接近(约8 min),比甲醇-水(72∶28,)作流动相时的保留时间(17.6 min)缩短了将近10 min。其次,乙腈的毒性大,成本较甲醇高,且流动相和提取溶剂种类一致能减小溶剂效应,因为实验中不同基质样品的提取溶剂均采用了甲醇,故综合考虑,选择相对低毒、低成本、且与样品基质分离效果良好的甲醇-水(80∶20)作为流动相。艾地苯醌标准溶液色谱图如图1所示。

图1 艾地苯醌标准溶液高效液相色谱图
2.4 检测波长的选择
取艾地苯醌标准溶液,以二极管阵列检测器,在190~400 nm 波长范围内进行扫描,得到艾地苯醌标准溶液的紫外吸收光谱图,如图2 所示。结果显示艾地苯醌在278 nm 附近有最大吸收。结合相关参考文献[12-14],选择278 nm 作为艾地苯醌的检测波长。
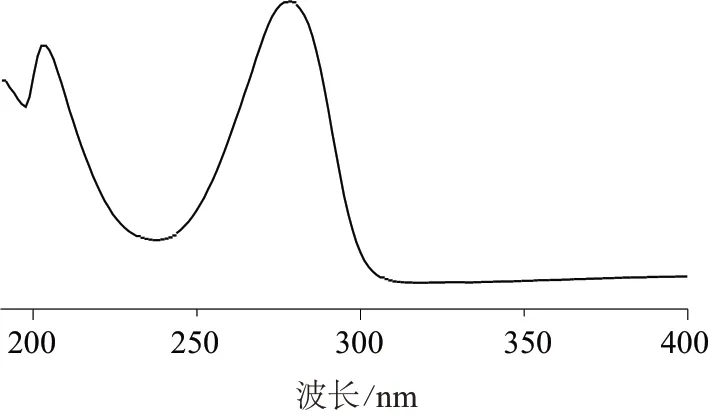
图2 艾地苯醌标准溶液紫外吸收光谱图
2.5 进样体积的选择
分别考察了进样体积为5、10、20 μL 时对测试结果的影响。结果显示,当进样体积为5 μL 时,信噪比过小影响检测精度;当进样体积为10 μL时,信噪比能达到要求,且色谱峰形较理想;当进样体积为20 μL、进样质量浓度达到1 000 mg/L 时,色谱柱出现样品过载,色谱图中出现拖尾峰。综合考虑,选择艾地苯醌的进样体积为10 μL。
2.6 线性关系、检出限及定量限
精密吸取艾地苯醌系列标准工作溶液各10 μL,注入液相色谱仪测定。以目标物的质量浓度(x,mg/L)为横坐标,色谱峰面积(y)为纵坐标,绘制标准工作曲线,计算线性回归方程和相关系数。
取膏霜、乳液、面膜、凝胶、液体水基5种空白样品各0.5 g,分别定量添加艾地苯醌标准溶液进行检测。在5种基质中,以信噪比为3时对应的溶液质量浓度确定方法检出限,以信噪比为10时对应的溶液质量浓度确定方法定量限。
艾地苯醌线性范围、回归方程、相关系数、方法检出限和定量限见表2。由表2可知,艾地苯醌质量浓度在1~200 mg/L范围内与对应色谱峰面积线性良好,相关系数为0.999 9,方法检出限为1.0 mg/kg,定量限为4.0 mg/kg。

表2 艾地苯醌的线性范围、线性方程、相关系数、检出限、定量限
2.7 加标回收试验
称取膏霜、乳液、面膜、凝胶、液体水基5种空白样品各0.5 g,分别加入适量艾地苯醌标准工作溶液,按1.3 方法处理,制成质量浓度分别为0.2、10、50 mg/L 的加标样品溶液,每一加标水平平行制备样品6 份,按1.4 色谱条件测定,计算加标回收率及其相对标准偏差(RSD),测定和计算结果见表3。由表3可知,艾地苯醌在5种化妆品基质中的平均加标回收率为95.61%~107.87%,测定结果的相对标准偏差为0.22%~3.07%,表明该方法精密度、准确度均较高,可满足日常化妆品中艾地苯醌的检测要求。5 种化妆品空白基质溶液、加标样品溶液色谱图见图3~图7。

图3 膏霜类化妆品空白基质、加标样品色谱图

图4 乳液类化妆品空白基质、加标样品色谱图

图5 面膜类化妆品空白基质、加标样品色谱图

图6 液体水基类化妆品空白基质、加标样品色谱图

图7 凝胶类化妆品空白基质、加标样品色谱图
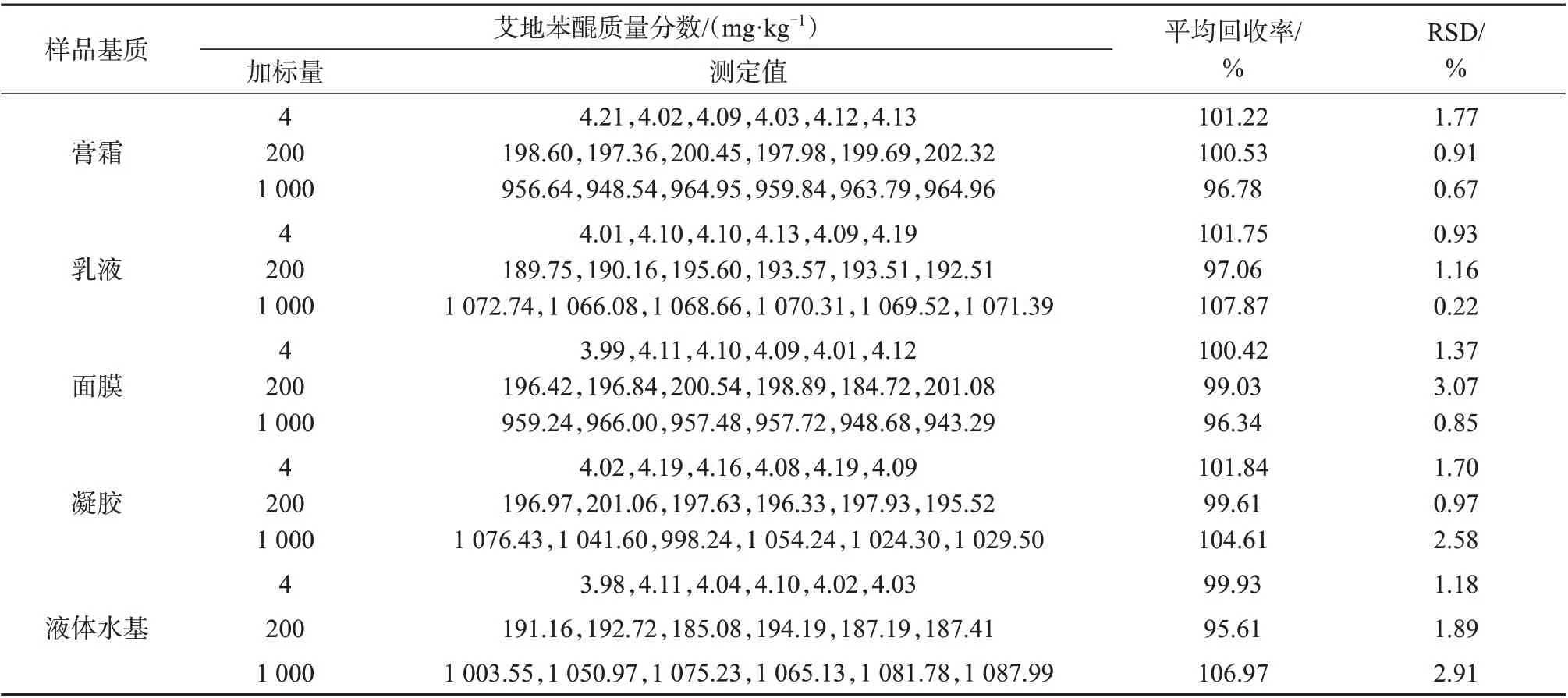
表3 空白样品加标回收试验结果
2.8 稳定性试验
分别精密吸取1、10、50 mg/L 3个浓度水平的膏霜、乳液、面膜、液体、凝胶水基5种空白样品的加标溶液各10 μL,分别于0、3、6、12、18、24、48、72 h 各进样一次,在1.4仪器工作条件下,记录不同时间的色谱峰面积并计算测定值的相对标准偏差,试验结果见表5。

表5 稳定性试验结果
由表5可知,5种空白样品的加标溶液在72 h内测得的艾地苯醌色谱峰面积的相对标准偏差为0.56%~2.32%,表明测试样品制备后在72 h内具有良好的稳定性。
3 结语
建立了高效液相色谱测定膏霜、乳液、面膜、凝胶、液体水基5 种基质类型的化妆品中艾地苯醌含量的分析方法。该方法具有操作简便、分析时间短、灵敏度高、精密度好、稳定性强、准确度高等优点,适用于功效化妆品中艾地苯醌的定性及定量检测,为打击化妆品监管中概念性添加及虚假宣称的违法行为提供一定的技术支持。
