溴结构域蛋白4 BD1和BD2选择性抑制剂的研究进展
2023-10-24黄菲王志杰卢帅陈亚东陆涛
黄菲,王志杰,卢帅,陈亚东,陆涛
(中国药科大学理学院 江苏 南京 211198)
1 概述
1.1 背景介绍
组蛋白的翻译后修饰(post-translational modifications,PTMs)是一种重要的表观遗传学修饰,参与细胞生命活动和疾病的发生发展进程。组蛋白PTMs目前已经报道出12种方式[1],包括组蛋白末端赖氨酸残基的乙酰化、甲基化、泛素化、磷酸化和磺酰化等。迄今为止,已在4个核心组蛋白(H2A、H2B、H3和H4)的N末端尾部和30个组蛋白变体中鉴定出130个修饰位点[2]。PTMs修饰会改变染色质的开闭程度,从而影响后续转录和基因表达过程。组蛋白赖氨酸的乙酰化是组蛋白翻译后修饰的重要方式之一[3],赖氨酸乙酰化在人类细胞中的频繁发生表明赖氨酸乙酰化在细胞信号传导网络中起着重要作用[4]。目前已有多个靶向组蛋白乙酰化过程的药物上市或在研,如靶向组蛋白去乙酰化酶(histone deacetylase,HDAC)的药物伏立诺他和贝利司他均已获批上市,分别用于治疗皮肤T细胞淋巴癌和多发性骨髓瘤[5]。
溴结构域蛋白(bromodomain-containing proteins,BCPs)可以特异地识别并结合组蛋白乙酰化的赖氨酸。目前已经在人类基因组中发现了61种BCPs,根据结构序列的相似性可分为8个BCPs家族,根据结构功能的不同又可分为溴结构域和超末端结构域家族(bromodomain and extra-terminal domain,BET)以及非溴结构域和超末端结构域家族(non bromodomain and extra-terminal domain,non-BET)。其中,BET家族包括溴结构域蛋白2、溴结构域蛋白3、溴结构域蛋白4和溴结构域蛋白T。正常情况下,位于细胞核内的BET蛋白能识别组蛋白H3和组蛋白H4中的乙酰化赖氨酸,招募转录调节复合物到染色质,参与以DNA为中心的转录调节过程中不同基因的表达[6]。BRD2、BRD3和BRD4在多种组织中表达,而具有高度组织特异性溴结构域蛋白T仅在粗线精母细胞、二倍体精母细胞和圆形精母细胞中表达[7]。作为BET家族成员之一,BRD4的功能已被发现与多种疾病的发生发展密切相关,尤其是与肿瘤的靶向治疗密切相关[8]。
1.2 BRD4的结构与功能
BRD4最初被命名为有丝分裂染色体相关蛋白(mitotic chromosomal-associated protein, MCAP),也被称为Fshrg4或Hunk1。BRD4蛋白有3种不同长度的异构体:1种长异构体(1 362个残基)和2种短异构体(分别为722和796个残基)[9]。在结构上,BRD4蛋白包括2个高度保守(氨基酸序列同源性高达95%)的N端溴结构域(BD1和BD2)、1个超末端结构域(extra-terminal,ET)和1个C端结构域(C-terminal domain,CTD);BD1和BD2能识别并结合乙酰化赖氨酸残基,ET结构域与辅因子结合发挥转录作用。BRD4的溴结构域分别由4个反向平行的α螺旋(αZ、αA、αB、αC)和连接螺旋的环状结构ZA loop和BC loop区域组成,ZA loop、BC loop 和αZ共同组成了WPF区域,αZ和αA组成了ZA channel区域,这些疏水间区域的相互作用形成了1个疏水性口袋,即乙酰化赖氨酸的结合口袋[10]。BRD4 BD1与组蛋白H4K8Ac12Ac的共晶复合物结构显示(见图1):乙酰化赖氨酸位于中心的疏水口袋中,与天冬酰胺Asn140形成关键氢键,而乙酰羰基的氧原子通过水分子与酪氨酸Tyr97产生氢键作用,同时乙酰化赖氨酸还与WPF区域和ZA channel区域产生疏水作用和静电相互作用。BRD4抑制剂能够通过竞争乙酰化赖氨酸与BRD4蛋白的结合位点来抑制BRD4蛋白的功能。

图1 BRD4 BD1与组蛋白H4K8Ac12Ac的共晶复合物(PDB ID:3UW9)Figure 1 Cocrystal structure of BRD4 BD1 with H4K8Ac12Ac (PDB ID:3UW9)
BRD4能够招募正性转录延长因子复合物(positive transcription elongation factor,P-TEFb),而该复合物在真核生物RNA聚合酶Ⅱ(RNA polymeraseⅡ,RNA PolⅡ)的转录调节中起重要作用[11],其功能失调常可导致多种人类疾病的发生发展。研究表明BRD4的功能紊乱与人类多种疾病如炎症、癌症、心血管疾病、中枢神经系统(central nervous system,CNS)疾病和人类免疫缺陷病毒(human immunodeficiency virus,HIV)感染等密切相关[7-8]。因此,BET被认为是治疗各种人类疾病的潜在靶点,已成为BCPs中最具药物敏感性的蛋白质家族[12-13]。BRD4包括2个串联的溴结构域BD1和BD2,二者具有高度同源性,在结合位点的氨基酸序列同源性高达95%[14]。尽管BD1和BD2在底物结合位点的序列高度相似(见图2)[15],但它们能在H3和H4组蛋白尾部特异性识别不同的乙酰化底物[16]。据报道,BD2对乙酰化底物有更广泛的作用,而BD1仅能识别H4组蛋白尾部的乙酰化底物[17]。因此,鉴于2个溴结构域的不同作用,设计选择性作用于不同溴结构域的BRD4抑制剂,既可用于探究BD1和BD2在疾病表型中的重要意义,也可推动BRD4抑制剂的药物研究。
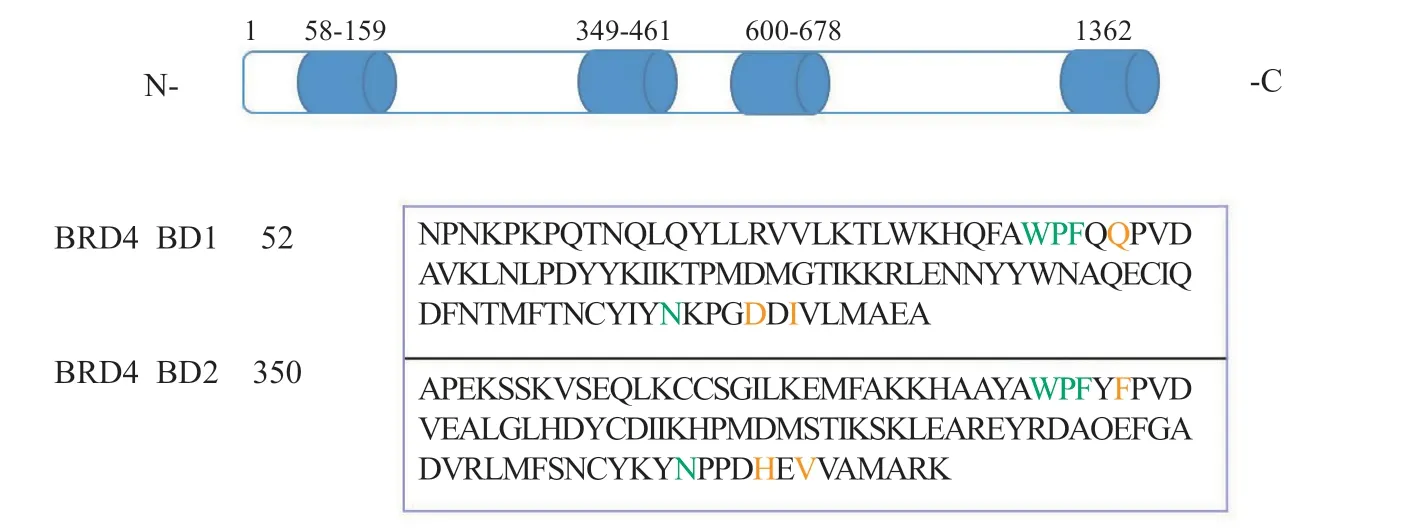
图2 BRD4 蛋白结构和BRD4 BD1与BRD4 BD2氨基酸序列对比Figure 2 BRD4 protein structure and conserved sequences of BRD4 BD1 and BRD4 BD2
2 BRD4抑制剂临床研究进展
尽管目前已有多个BRD4抑制剂进入临床研究(见表1),但进展缓慢,至今尚无药物上市。多项研究表明头晕、恶心和呕吐等不良反应严重影响了BRD4抑制剂的临床进展,甚至导致临床试验终止。而缺乏对BRD4蛋白BD1和BD2结构域的选择性可能是其中的原因之一[15]。选择性靶向BRD4 BD1或BRD4 BD2的抑制剂已成为目前研究的热点。本文综述了目前已经报道的选择性BRD4蛋白抑制剂,按照对不同结构域的选择性作用区分,包括选择性BRD4 BD1抑制剂和选择性BRD4 BD2抑制剂。

表1 进入临床研究的BRD4抑制剂Table 1 BRD4 inhibitors under clinical trials
2.1 BRD4 BD1选择性抑制剂
Raux等[18]通过高通量筛选发现,BRD4 BD1选择性抑制剂化合物1能够准确地结合到ZA channel中,与Asn140形成氢键,并通过水分子介导与Tyr97形成氢键,此外还与蛋白结合口袋底部保守的水分子网络连接起来。化合物1对BRD4 BD1的IC50达到微摩尔级别(IC50= 5 μmol · L-1),较BET家族其他成员的选择性高10倍。化合物1能剂量依赖地降低Jurkat T细胞活力,并且能下调原癌基因c-Myc的水平。
Rodriguez等[19]通过虚拟筛选得到的化合物2,是一种具有重氮苯结构的BRD4 BD1选择性抑制剂,其对BRD4 BD1的Ki为30 ~ 50 nmol · L-1,选择性是BRD4 BD2的10倍。随后发现的化合物3是一种具有环丙烯结构的BRD4 BD1选择性抑制剂,对BRD4 BD1的Ki为77 nmol · L-1,较BRD4 BD2的选择性超过9倍。从化合物4与CBP蛋白结合的作用机制出发,他们又发现了一种四氢吡啶吲哚结构的化合物5(olinone)[14],该化合物对BRD4 BD1的Kd为3.4 μmol · L-1,对BRD4 BD2的选择性超过100倍,对BRD2、BRD3、BRD4的BD1均具有一定的选择性。化合物5与BRD4 BD1蛋白晶体复合物结构表明,Olinone 中乙酰基的羰基氧与BRD4 BD1的Asn140酰胺键的氮原子形成氢键,三杂环结构与BD1乙酰赖氨酸的结合口袋处的Asp144形成了氢键,而在BD2蛋白中该位置是His437残基,该位点在BET家族中并不是保守的,这也解释了olinone对BET家族中的其他BD1蛋白产生选择性的原因。Olinone能够促进少突胶质细胞祖细胞分化,而非选择性抑制剂则会抑制少突胶质细胞祖细胞分化,BRD4 BD1选择性抑制剂或许能成为未来治疗脱髓鞘疾病的一种新策略。
中南大学湘雅医学院的研究者通过对BRD4 BD1和BRD4 BD2结合口袋处的叠合共晶结构分析[20],发现二者主要的结构差异在于BC loop中的Asp144(BD1)和His437(BD2);BRD4 BD2口袋空腔比BRD4 BD1口袋的空腔窄,因此在非选择性BRD4抑制剂JQ1的基础上在BD2区域插入与His437形成位阻的结构基团,以达到对BD1的选择性抑制,最终得到了化合物6[20]。化合物6对BRD4 BD1的IC50为0.56 μmol · L-1,而对BRD4 BD2的IC50大于100 μmol · L-1,化合物6与BRD4 BD1和BRD4 BD2的叠合共晶结构表明,叔丁基的引入使得化合物6与His437(BD2)产生碰撞而无法与蛋白口袋结合,这是对BD2活性差的原因。化合物6与BRD4 BD1的共晶结构显示,化合物6的3,5-二甲基异恶唑在乙酰化赖氨酸口袋处与Asn140结合形成氢键,WPF区域3,4-二氯取代基的引入使3,4-二氯苯基的平面趋于垂直于嘧啶酮的平面,嘧啶酮可能与Trp81产生了π-π堆积作用。化合物6对肺成纤维细胞系(HLF-1)、肾成纤维细胞系(NRK-49F)和肝成纤维细胞系(LX2)的IC50为4.2 ~ 19.6 μmol · L-1。I型胶原是细胞外基质(extra cellular matrix,ECM)的主要成分,也是控制纤维化过程的关键因素,化合物6能剂量依赖性地抑制Ⅰ型胶原的表达,并且在相同浓度的10 μmol · L-1下,化合物6比(+)-JQ1更有效,而RVX-208对Ⅰ型胶原的表达没有抑制作用,说明BRD4 BD1在纤维化过程中起主导作用,选择性抑制BRD4 BD1比同时抑制BRD4 BD1和BRD4 BD2具有更强的抗纤维化活性。化合物6可以作为一种新的先导化合物,进一步开发针对器官纤维化或癌症的有效选择性BRD4 BD1抑制剂。

美国德克萨斯大学研究者发现的化合物7(ZL0513)和化合物8(ZL0516)[21]是根据RVX-208和RVX-OH与BRD4 BD1在WPF区域的不同结合模式,基于配体结构的药物设计与骨架跃迁策略,设计出的具有香豆素母核结构的系列化合物,化合物ZL0513对BRD4 BD1的IC50为67 nmol · L-1;ZL0516对BRD4 BD1的IC50为84 nmol · L-1,对BRD4 BD2的选择性大于10倍(BRD4 BD2 IC50=718 nmol · L-1),且对BET家族其他成员也具有较高的选择性[21],化合物8能准确地结合到乙酰化赖氨酸口袋中(见图3),结构中手性碳上的羟基通过一个水分子介导与BRD4 BD1特有的Asn93形成氢键,羰基与Asn140相互作用,香豆素母核中的一个甲氧基通过水分子介导与Tyr97形成氢键,同时被包裹在乙酰化赖氨酸口袋中,另一个甲氧基则延伸到WPF区中。ZL0516可用于治疗慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD),ZL0516具有良好的成药性,其生物利用度(F)大于35%,水溶性和和代谢稳定性较好,对心脏的毒性和肝药酶CYP450的抑制较弱,有望作为先导化合物继续优化,推向进一步临床研究。该团队研发的化合物9(ZL0590)[22]以一个在此之前从未报道过的结合方式选择性作用于BRD4 BD1蛋白,蛋白晶体复合物结构显示,ZL0590与αB和αC螺旋上的非乙酰化赖氨酸口袋位点结合,作用于BRD4 BD1上特异的氨基酸残基,吗啉环的碱性胺部分可被质子化,并与Glu151的羧酸部分形成盐桥,磺酰胺与Gly143的主链酰胺形成氢键,磺酰胺中的另一个氧原子与水分子形成氢键,从而介导与Asp144主链羰基的相互作用,尿素连接部位通过2个水分子与Tyr137的主链羰基和Glu154的侧链羧酸基团相互作用。化合物ZL0590对BRD4 BD1的IC50为90 nmol · L-1,对BRD4 BD2的选择性大于10倍(BRD4 BD2 IC50= 1 093 nmol · L-1),体外代谢实验表明,ZL0590的生物利用度较为良好(F = 24%),半衰期达到3.7 h,药时曲线下面积为602 ng · h · mL-1,最大浓度为86.9 ng · mL-1,水溶性为3 500 μg · mL-1。体内药效实验表明,ZL0590能够有效抑制poly(I:C)诱导的小鼠肺组织中性粒细胞炎症,降低炎症因子白细胞介素6(interleukin-6,IL-6)等的表达,用于治疗COPD。该化合物这一新颖结合蛋白模式的发现,将为今后阐明BRD4选择性抑制剂的复杂机制和BRD4蛋白功能奠定重要基础。

图3 化合物8与BRD4 BD1蛋白共晶结合模式图(PDB ID:6UWU)Figure 3 Cocrystal structure of compound 8 with BRD4 BD1 (PDB ID: 6UWU)
郑州大学药学院的研究者们发现,具有茶碱母核的一系列化合物能够选择性抑制BRD4 BD1蛋白,经过一系列基于片段的结构优化,得到化合物10(BRD4 BD1 IC50= 2.51 μmol · L-1),对BRD4 BD2的选择性大于20倍[23]。分子对接模型显示该系列化合物都能和BD1蛋白结合口袋的Asn140产生氢键相互作用,化合物10产生最佳选择性的原因可能是N-7位置的苯基与BRD4 BD1蛋白的Trp81氨基酸残基形成T形π-π堆积相互作用,同时与BRD4 BD1蛋白中的大体积氨基酸残基Ile146形成紧密π-π相互作用,而BRD4 BD2蛋白中该位置则是体积稍小的Val439。

2020年明尼苏达大学的研究者从自己前期设计的具有微摩尔活性的泛BD1抑制剂11出发,该化合物同时对丝裂原活化蛋白激酶p38α(mitagenactivated protein kinase p38α,MAP p38α)也有一定的抑制作用(Kd= 0.47 nmol · L-1),基于结构优化得到化合物12,其对BRD4 BD1的IC50为0.13 μmol · L-1,对BET其他家族成员选择性为9 ~ 33倍[24]。2022年Cui等[25]从化合物12(UMN627)对BRD4 BD1产生选择性的机制出发,UMN627具有对BRD4 BD1独特选择性的原因之一是化合物在与BD1蛋白结合位点占据了3个水分子的位置。在此结构基础上进一步进行结构优化,得到了化合物13。化合物13对BRD4 BD1的IC50为92 nmol · L-1,对BRD4 BD1的Kd为18 nmol · L-1,对BRD4 BD1的选择性相对于BRD2 BD1和BRD4 BD2都大于500倍。蛋白共晶结构显示(见图4),化合物13结构中的咪唑环与BRD4 BD1的Asn140形成氢键,三氟甲基苯基取代了BRD4 BD1蛋白结构中的3个水分子,二甲基乙氨基通过水分子与Asn140和Asp144形成氢键,这是化合物13产生特异选择性的结构基础。化合物13能够影响白血病细胞MM.1S内BRD4蛋白的表达,但在低浓度下未观察到对原癌基因c-Myc的抑制,而非选择性抑制剂在低浓度下则会产生对原癌基因c-Myc的抑制。化合物13能够降低非小细胞肺癌细胞系中的细胞因子IL-8的水平,并能在人肝窦内皮细胞中抑制炎症导致的趋化因子表达。

图4 化合物13与BRD4 BD1蛋白共晶结合模式图(PDB ID:6WGX)Figure 4 Cocrystal structure of 13 with BRD4 BD1 (PDB ID:6WGX)
Cui等[26]在设计出UMN627之后,在化合物14的结构基础上,根据ABBV-744的构效关系,改变三氮唑环上的芳基,可以增强对BD1的选择性,发现了化合物15(DW34),DW34对BET家族的BD1有选择性,对BETs BD1的Kd为12 ~ 22 nmol · L-1,对BD2的选择性大于16倍(对BRD2 BD2、BRD3 BD2、BRDT BD2的Kd为200 ~ 310 nmol · L-1),而对BRD4 BD2的Kd为67 nmol · L-1。晶体结构表明DW34结构的二甲基乙氨基与BD1中特有的Asp144通过一个水分子介导形成了氢键(见图5),GSK778也产生了类似的氢键作用,DW34对BRD4 BD2有选择性的原因至今尚不明确。此外,DW34能显著抑制原癌基因c-Myc的表达,EC50<100 nmol · L-1,同时能够抑制白血病细胞MM.1S的生长和下调炎症细胞因子IL-8水平。
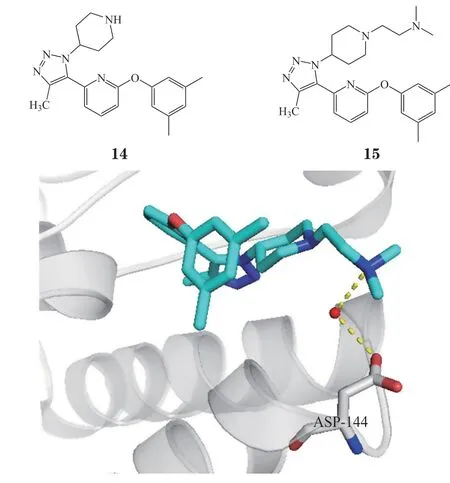
图5 化合物15与BRD4 BD1蛋白共晶结合模式图Figure 5 Cocrystal structure of compound 15 with BRD4 BD1
化合物16(GSK778)[27]是葛兰素史克公司根据I-BET151的结构研发的泛BD1选择性抑制剂(IC50= 40 nmol · L-1),对BD2的选择性大于130倍,化合物16与BRD4 BD1共晶结构表明其产生选择性的基础是结构中的四氢吡咯环与BD1蛋白中特有的Asp144通过水分子介导形成的氢键,Asp144侧链受到Lys141氢键的限制,而在BD2蛋白中该位置是氨基酸残基His433和Pro430。一些细胞实验表明GSK778与非选择性BET抑制剂I-BET151的作用相似,能够抑制人白血病细胞K562增殖,诱导细胞周期阻滞和凋亡,能降低急性髓细胞白血病(acute myelocytic leukemia,AML)细胞的克隆形成能力。在GSK778的基础上进一步进行结构优化,得到的化合物17(GSK789),其具有更高的BD1选择性(BRD4 BD1 IC50= 20 nmol · L-1),对BRD4 BD2的选择性达到1 500倍(BRD4 BD2 IC50= 30 μmol · L-1)。与GSK778产生选择性的基础类似,其与BD1蛋白中的Asp144通过水分子介导形成氢键,同时结构中的呋喃环与ZA channel中的Trp81、Phe82和Leu92侧链形成紧密的疏水作用,GSK789在一系列癌细胞系中显示出强大的抗增殖作用,具有一系列抗增殖、抗炎、免疫调节和组织重塑活性。但GSK789在大鼠和小鼠肝细胞中的清除率较高,因此没有进行体内代谢研究。

笔者所在课题组在前期基于片段的筛选得到苯并[cd]吲哚-2-(1H)-酮类化合物18,进一步对其与BRD4 BD1蛋白不同的结合区进行结构优化得到化合物19(LT052)[28],其对BRD4 BD1的IC50为87.7 nmol · L-1,对BRD4 BD2的选择性为138倍(BRD4 BD2 IC50= 12.1 μmol · L-1)。化合物19与BRD4 BD1的晶体复合物显示,该化合物结构中的甲基咪唑环与BD2蛋白中的His437残基产生了空间位阻,阻碍了其与BD2蛋白的结合,从而增加了对BD1的选择性。在体外评估实验中,LT052能够减少小鼠单核巨噬细胞白血病细胞RAW264.7中NO的产生,而BD2抑制剂RVX-208抑制NO产生的效果不如LT052,同时LT052能够下调核因子κB(nuclear factor kappa-B,NF-κB)通路的信号传导,且效果优于(+)-JQ1。体外肝微粒体实验研究表明化合物19在大鼠、小鼠、猴子和人肝微粒体中的半衰期为9.4 ~ 14.8 min,清除率较高,为93.517 ~146.685 μL · min-1· mg-1,LT052能通过抑制BRD4影响NF-κB/NOD样受体热蛋白结构域相关蛋白3(NOD-like receptor thermal protein domain associated protein 3,NLRP3)信号通路,抑制氧自由基清除剂尿酸钠(monosodium urate,MSU)诱导的巨噬细胞THP-1细胞炎症级联反应和细胞焦亡。在大鼠动物模型中,LT052通过调节该信号通路抑制大鼠滑膜组织中巨噬细胞的焦磷酸化,从而实现急性痛风性关节炎的治疗。

2.2 BRD4 BD2选择性抑制剂
化合物20(GSK046)[29]是葛兰素史克公司通过高通量筛选得到的BD2选择性抑制剂。GSK046与BRD4 BD2的共晶结构显示(见图6),GSK046结构中的苄基和环己酰胺与BRD4 BD2蛋白中的Pro430和His433形成广泛的疏水作用力,而在BD1蛋白中这些位置被Asp144和Lys141取代,这是其产生选择性的基础。为了改善成药性,并保留GSK046对BD2的特异选择性,设计合成了化合物21(GSK620),GSK620在胶原诱导的大鼠关节炎模型中能够剂量依赖地降低免疫球蛋白IgG1水平,显著减少关节肿胀、滑膜炎、软骨损伤、血管翳和骨吸收等症状,在咪喹莫特(imiquimod,IMQ)诱导的银屑病小鼠模型中减少红斑和斑块形成和表皮增生方面的效果优于近期美国FDA批准的治疗银屑病的阿普司特,并显著降低了与疾病相关的促炎症基因IL-17A、IL-17F和IL-22的表达,在非酒精性脂肪性肝病(nonalcoholic fatty liver disease,NAFLD)小鼠模型中,与对照组相比,GSK620治疗组小鼠的脂肪变性、小叶炎症和肝细胞气球化减少,能够减少肝活检中促炎症和促纤维化基因的表达。

图6 化合物GSK046与BRD4 BD1/BRD4 BD2的蛋白共晶叠合图(PDB ID:6SWQ, 6SWP)Figure 6 Cocrystal structure of compound 20 with BRD4 BD1/BRD4 BD2 (PDB ID: 6SWQ,6SWP)
在泛BRD4抑制剂22(ABBV-075)的基础上,研究人员基于结构优化策略得到的选择性BD2抑制剂23(ABBV-744),其对BRD4 BD1的IC50为27.5 nmol · L-1,对BRD4 BD2的IC50为20.7 μmol · L-1。化合物ABBV-744目前已经进入Ⅰ期临床试验阶段[30],用于治疗前列腺癌和多发性骨髓瘤。在ABBV-744分别与BRD4 BD2和BRD4 BD1的晶体复合物结构中发现,ABBV-744维持了ABBV-075所有重要的相互作用。ABBV-744的乙酰胺部分通过将酰胺埋在由BD2的His433、Tyr386和Pro430残基形成的通道中,ABBV-744的2,6-二甲基苯醚利用Ile162(BD1)和Val435(BD2)的细微体积大小差异,芳基甲基埋入WPF口袋的刚性底部,BD2中较小的Val435残基可以适应这种甲基相互作用,而对于BD1蛋白,这种芳基甲基与体积较大Ile162残基的相互作用迫使抑制剂稍微偏离Ile部分,这是对BD2产生选择性的结构基础。ABBV-744在前列腺癌细胞LNCaP小鼠异种移植模型中,在最高耐受剂量的1/12时,ABBV-744能够产生显著的抗肿瘤活性。而小鼠毒性研究中,ABBV-075经口给药剂量为3 mg · kg-1(LNCaP小鼠异种移植模型中有效暴露量的3倍),导致血小板减少59%,杯状细胞丢失。相比之下,30 mg · kg-1的ABBV-744(有效暴露量的25倍)导致血小板减少仅20%,并且60 mg · kg-1(有效暴露量的47倍)不会导致杯状细胞丢失或其他肠道缺陷。同样,2.5 mg · kg-1的ABBV-075导致睾丸中的生殖细胞变性,而25 mg · kg-1剂量下的ABBV-744未观察到睾丸中的变化。这些疗效和耐受性结果共同表明,选择性靶向BD2可以在某些癌症环境中诱导抗肿瘤活性,同时缓解非选择性BET抑制剂的关键耐受性问题。代谢实验研究表明,ABBV-744主要由CYP3A4代谢,在小鼠和狗体内的清除率较低,而在大鼠和猴子体内的清除率适中,t1/2为2.0 ~ 4.4 h,口服生物利用度为7% ~77%,具有较好的成药性。

南京中医药大学的研究者从中药材金合欢属植物大叶相思和坚叶相思树干中提取得到的3,4,7,8-四羟基黄酮天然产物化合物24[31],与其他已经报道的BRD4抑制剂相比,这是BRD4选择性抑制剂中一个新颖的化学骨架。化合物24能够选择性抑制BRD4 BD2蛋白(BRD4 BD2 IC50= 204 nmol · L-1),其对BRD4 BD1的选择性大于100倍(BRD4 BD1 IC50= 17.9 μmol · L-1)。化合物24与BRD4 BD1和BRD4 BD2的2个共晶结构都在相似的位置形成了3个氢键。在BRD4 BD2中,化合物24与Asn433和Tyr390附近的水分子形成2个氢键作用力,与Pro375的主链羰基形成第3个氢键;而在BRD4 BD1中,化合物24则分别和Asn140、Tyr97和Pro82形成氢键。3个氢键将化合物24的香豆素母核牢牢地固定在乙酰化赖氨酸结合口袋中,此外,疏水残基Val87、I146和Ile94(BRD4 BD1)或Val380、Val439和Lys387(BRD4 BD2)与化合物24形成强烈的疏水相互作用,在与BD2的共晶结构中,化合物24的苯基的电子云密度比与BD1的电子云密度更低,显示了更强的相互作用和更稳固的空间构象,化合物24与BD2结构中的Asn433附近的水分子形成了1个氢键,同时可能与His437形成疏水相互作用;而在BD1该位置中对应的残基是氨基酸残基Asp144,其侧链距离化合物24太远,无法形成有效的相互作用。化合物24与非选择性BRD4抑制剂RVX-208的母核结构相似,但共晶复合物结构显示两者的结合模式截然不同。体内外药效实验表明,化合物24能够在mRNA水平和蛋白质水平显著降低原癌基因c-Myc的表达;在小鼠异种移植MV4-11肿瘤模型中,在50 mg · kg-1剂量下,肿瘤的大小明显小于阴性对照组,在100 mg · kg-1剂量下,肿瘤的大小与50 mg · kg-1的(+)-JQ1治疗组相当,且毒性较低。该化合物作为一个新颖的化学骨架,能够作为一个良好的先导化合物来开展BRD4 BD2选择性抑制剂的研究。
RVX-208(25)[32]是泛BD2抑制剂,目前处于Ⅲ期临床试验,用于治疗与动脉粥样硬化相关的心血管疾病,AlphaScreen方法测定其对BRD4 BD1的IC50为87 μmol · L-1,而对BRD4 BD2的IC50为0.510 μmol · L-1,选择性为170倍,对于BRD3的BD2和BD1,Kd分别为195和4 μmol · L-1。RVX-208与BRD2 BD2蛋白的共晶结构表明RVX-208结构上的喹唑啉酮环系统的羰基氧和1个氮原子充当乙酰赖氨酸模拟部分,与保守的天冬氨酸Asn140形成氢键,同时通过1个水分子与Tyr97形成氢键,RVX-208占据单个乙酰赖氨酸使用的整个通道,羟基乙醚部分从乙酰赖氨酸结合袋中伸出,BRD2中的BD2独特残基His433翻转到乙酰赖氨酸结合位点,与RVX-208的苯环结合,与BRD4 BD2结构域有更紧密的亲和力,与BRD2 BD2蛋白表面接触面积较少。然而,RVX-208与BRD4 BD1特有的残基几乎没有直接相互作用,仅仅形成了与Gln85的水介导氢键,这解释了RVX-208对BD2产生选择性的原因。

3 结语与展望
目前,泛BRD4抑制剂由于不良反应等问题在临床研究中进展缓慢,开发选择性BRD4抑制剂成为科研上亟待攻克的难题。而新一代的高选择性BRD4 BD1或BD2抑制剂因其具有较好的安全性等优势,取代泛BRD4抑制剂成为开发选择性BRD4抑制剂的趋势。本文综述了近年来报道过的对BRD4蛋白的BD1选择性抑制剂和BD2选择性抑制剂,产生选择性的基础在于化合物分子在不同的溴结构域上与其独有的氨基酸残基产生相互作用,对BRD4 BD1产生选择性的结构基础包括BD1结合口袋处的Asp144和产生堆积作用力的Asn93等关键氨基酸残基,对BRD4 BD2产生选择性的结构基础包括BD2结合口袋附近的Pro430、His433、Pro375和Tyr97等关键氨基酸残基。同时天然产物分子化合物24特殊的骨架能产生对BRD4 BD2良好的选择性,这为今后研究出高选择性的BRD4 BD2抑制剂提供了思路,从天然产物中开发高选择性BET抑制剂不失为一种良好的策略。目前已经报道过的大部分选择性抑制剂均是作用于乙酰赖氨酸的结合口袋处,通过竞争乙酰化赖氨酸来抑制BRD4蛋白的表达,而化合物ZL0590则作用于非乙酰化赖氨酸区域,以一种独特的方式作用在BRD4蛋白内部,同时能够与BD1和BD2产生不同的作用,这打破了研究者的思路,即开发BRD4抑制剂不一定必须作用于乙酰化赖氨酸结合口袋,这也开启了今后开发选择性BET抑制剂新的大门。
选择性BD1或BD2抑制剂已表现出毒性低、特异性强的优点。脱髓鞘疾病目前无有效根治措施,主要采取对症和支持疗法,BRD4 BD1选择性抑制剂olinone能够促进少突胶质细胞祖细胞分化,或许这是未来脱髓鞘疾病的一种较有前景的治疗方法。BRD4 BD1选择性抑制剂RVX-208具有独特的抗纤维化活性,今后可能在治疗纤维化疾病中能发挥重要作用。多种BRD4 BD1选择性抑制剂还在炎症反应中具有良好的治疗优势,能够下调多种炎症因子水平,具有治疗关节炎和COPD等炎症因子导致疾病的潜力。BRD4 BD2选择性抑制剂GSK620对银屑病和非脂肪性肝炎具有良好的效果,BRD4 BD2选择性抑制剂ABV-744则对前列腺癌有较好的作用。根据已经报道的选择性抑制剂,均表现出对多种肿瘤特异性强且耐受性高的效果,未来继续开发具有选择性的BRD4 BD1或BD2抑制剂或许能为许多尚未攻克的疾病如癌症提供治疗思路,解决人类在疾病发生发展过程中的难题。
