N6-甲基腺苷去甲基化酶小分子抑制剂的研究进展
2023-10-24汪香涵徐俊费文龙尤启冬郭小可
汪香涵 ,徐俊,费文龙 ,尤启冬 *,郭小可 **
(1.中国药科大学 江苏省药物分子设计与成药性优化重点实验室,江苏 南京,211198;2.中国药科大学药学院,江苏 南京,211198)
表观遗传学主要针对在基因核苷酸序列不发生改变的情况下,对基因表达的可遗传变化进行探究,主要涉及的机制包括DNA修饰、组蛋白修饰和RNA编辑修饰等[1]。RNA的动态修饰对生命体的多种生理过程具有至关重要的作用,如细胞周期调控、细胞增殖分化等。真核生物的RNA修饰位点主要包括N6-甲基腺苷(N6-methyladenosine,m6A)、5-甲基胞嘧啶(5-methylcytosine,m5C)以及N1-甲基腺苷(N1-methyladenosine,m1A)等。其中m6A是信使RNA(message RNA,mRNA)修饰中最常见、最保守,同时也是目前RNA修饰研究的热点之一[2]。哺乳动物细胞中的m6A修饰具有可逆性,其修饰过程如图1所示。m6A的识别由m6A阅读蛋白完成,而其甲基化修饰和去甲基化修饰则是由m6A甲基转移酶和m6A去甲基化酶参与调控。
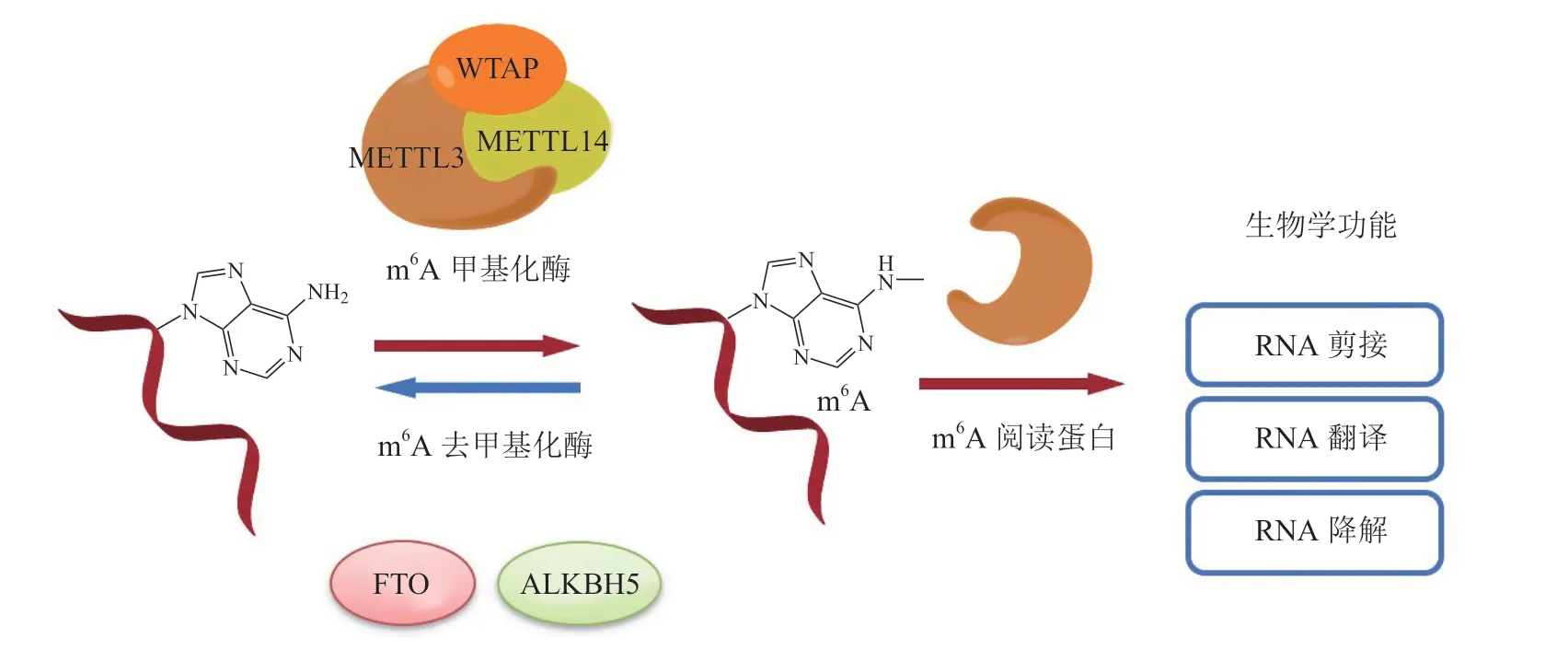
图1 m6A的动态修饰Figure 1 Dynamic modification of m6A
2012年以前,由于缺乏具体的检测手段,m6A在生物体内的分布和作用并未明确。2012年,Dominissini等[3]发明了基于m6A的RNA甲基化免疫沉淀测序方法(methylated RNA immunoprecipitation sequencing,MeRIP-Seq),从此逐渐揭开了m6A的神秘面纱。m6A甲基化由甲基转移酶复合体催化。甲基转移酶复合体是由若干RNA甲基转移蛋白组合而成,分别是S-腺苷甲硫氨酸(S-adenosylmethionine,SAM)结合蛋白——甲基转移酶样蛋白3(methyltransferase-like protein 3,METTL3)、甲基转移酶样蛋白14(methyltransferase-like protein 14,METTL14)和WT1-相关蛋白(WT1-associated protein,WTAP)[4]。其中METTL3起到催化作用,其内源SAM结合口袋可将SAM上的甲基转移至腺苷形成腺苷半胱氨酸;而METTL14的作用是稳定甲基转移酶复合体并识别特异性RNA作为催化底物;WTAP没有催化作用,但它可招募METTL3和METTL14进入细胞核内。当腺苷被甲基化后,这种化学修饰则被阅读蛋白识别,影响后续RNA的剪切、转录以及降解。m6A阅读蛋白主要包含YT521-B同源结构域(YT521-B homology domain,YTH domain)的YTH家族蛋白以及其他潜在可与m6A结合的蛋白。它们已经被证实可以通过募集不同的复合物靶向m6A修饰位点来参与转录后的基因调控。
2011年,Jia等[5]首次发现了FTO可催化核内mRNA的去甲基化,2年后发现同蛋白家族的ALKBH5也是mRNA去甲基化酶[6]。近期有研究发现,另一同源蛋白ALKBH3对tRNA上的m6A也有去甲基化活性[7],它们均属于Alkb同源蛋白家族。Alkb同源蛋白在哺乳动物中存在9种亚型,分别是ALKBH1 ~ ALKBH8和FTO(ALKBH9)。其中ALKBH2和ALKBH3是核DNA修复酶[8];ALKBH3同时还是tRNA的去甲基化酶,作用位点为tRNA上的m1A、m3C和m6A[7];而ALKBH5和FTO则是mRNA去甲基化酶。ALKBH8是tRNA修饰蛋白酶,可将tRNA中的5-羧甲基尿苷(5-carboxymethyluridine,cm5U)甲基化成5-甲氧羰基甲基尿苷(5-methoxycarbonylmethyluridine,mcm5U)[9]。随后羟基化成(S)-5-甲氧羰基羟甲基尿苷[(S)-5-methoxycarbonylhydroxymethyluridine,S-mchm5U][10]。ALKBH1的生物学功能众说纷纭,目前已经发现的包括DNA、tRNA、mRNA和组蛋白的去甲基化作用[11]。最近还发现其对碱基未正确配对的双链DNA上的m6A有着较高的催化活性[12]。ALKBH4与肌动蛋白的去甲基化有关[13]。最近发现它还具有修复DNA m6A的功能[14]。ALKBH7是一种线粒体蛋白,与细胞的程序化凋亡有关[15]。而ALKBH6的相关生物学作用机制研究鲜有报道,尚需深入探究。
2012年,Chen等[16]报道了首个FTO小分子抑制剂——大黄酸;随后又发现了FTO的选择性抑制剂甲氯灭酸[17];后续结构优化得到的活性更好的小分子抑制剂FB-23,为FTO的生物学机制研究提供帮助[18]。近年来,已经有多项研究证明m6A的异常丰度与许多恶性肿瘤有着密切的关系,如乳腺癌、肺癌、胃癌以及急性髓系白血病等[19-20]。因此m6A被许多学者认为是潜在的肿瘤治疗靶点,对肿瘤的治疗有着重要的理论依据和应用价值。本文针对m6A去甲基化酶从相关癌症和小分子抑制剂研究进展等方面进行综述,为相关领域药物的进一步研发提供参考。
1 m6A去甲基化酶
1.1 m6A去甲基化酶FTO
最初FTO是作为肥胖相关蛋白被人们关注,与成人早发性肥胖和重度肥胖有关。随着RNA表观遗传学研究的逐步深入,人们发现m6A参与到RNA的生理活动中,并且FTO能对m6A进行去甲基化修饰,在多种肿瘤调控中发挥重要作用。FTO属于Fe2+和α-酮戊二酸(2-oxoglutaric acid,2-OG)依赖的加氧酶,能够将m6A中的N-甲基氧化成羟甲基,形成N6-羟甲基腺嘌呤(N6-hydroxymethyladenosine,hm6A),羟甲基不稳定,自发脱水失去1分子甲醛,得到腺嘌呤结构(见图2)。FTO发挥作用依赖2-OG和Fe2+,其中的关键氨基酸残基His307、His231和Asp233参与金属离子螯合,Glu234参与m6A底物识别过程,Try108参与RNA碱基识别[21](见图3)。
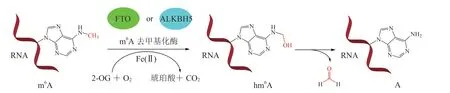
图2 m6A的去甲基化过程Figure 2 Demethylation of m6A
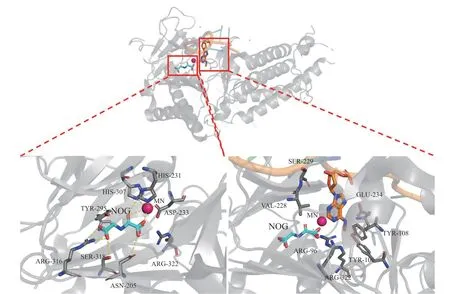
图3 m6A与FTO共晶复合物(PDB ID: 5ZMD)Figure 3 Cocrystal structure of FTO in complex with m6A(PDB ID: 5ZMD)
除了m6A外,FTO还有许多作用底物,如单链DNA中的N3-甲基胸腺嘧啶(N3-methylthymidine,m3T)和RNA中的N3-甲基尿嘧啶(N3-methyluridine,m3U)[22]。FTO的催化底物与定位分布密切相关,FTO不仅存在于细胞核中,在某些情况下也存在于细胞质中。在细胞核中FTO作用底物为多聚腺苷酸(polyA)RNA中的m6A、核小RNA(small nuclear RNA,snRNA)中的N6,2'-O-二甲基腺苷(N6,2'-O-dimethyladenosine,m6Am)、tRNA中的m1A;而在细胞质中则是polyA RNA中的m6A和m6Am、tRNA中的m1A[23]。最近,有研究指出FTO的最佳催化底物并非m6A,而是m6Am[24]。在FTO敲除的细胞中,m6A的总量变化不明显,而m6Am明显增加,这提示RNA上其他类型的甲基化修饰可能是FTO的主要作用底物。
随着FTO相关的表观遗传学研究深入,越来越多的证据表明FTO与肿瘤的产生与增殖分化有着密切关系。在某些恶性肿瘤中,FTO呈现高表达状态,可能影响肿瘤的增殖与分化,如急性髓细胞白血病[25]、乳腺癌[26]、肝癌[27]、黑色素瘤[28]、胰腺癌[29]、膀胱癌[30]和胃癌[31]等。2017年,Li等[25]报道了FTO作为m6A去甲基化酶在急性髓细胞白血病(acute myelocytic leukemia,AML)中的病理功能。FTO在AML的某些亚型,如携带MLL1基因异位混合细胞系白血病(mixed lineage Leukemia,MLL)中高度表达。研究表明,FTO作为m6A去甲基化酶,通过调控锚蛋白重复序列-细胞信号抑制因子盒蛋白家族2(ankyrin repeat and SOCS box containing 2,ASB2)和维甲酸受体α(retinoic acid receptor alpha,RARA) mRNA上m6A的丰度,抑制全反式维甲酸(all-trans-retinoicacid,ATRA)诱导的AML细胞分化并促使白血病的发生(见图4)。2018年Su等[32]发现化合物2R-羟基戊二酸(R-2-hydroxyglutarate,R-2HG)能抑制FTO蛋白的活性,使m6A的丰度增加,骨髓细胞瘤病毒癌基因同源物(myeloma viral oncogene homolog,MYC)和CCAAT增强子结合蛋白基因(CCAAT enhancerbinding protein alpha,CEBPA)mRNA稳定性下降,从而导致白血病细胞生长抑制、细胞周期阻滞和凋亡。2022年Zhou等[33]发现在T细胞急性淋巴细胞白血病(T-cell acute lymphoblastic leukemia,T-ALL)中,FTO升高可逆地降低了干扰素调节因子 8(interferon regulatory factor 8,IRF8)的m6A修饰,通过改变mRNA稳定性来降低IRF8表达,FTO抑制剂FB23-2有效降低了IRF8的表达并延长了T-ALL小鼠的存活期。因此,针对FTO靶点开发小分子抑制剂在未来是一种可行的AML治疗策略。
Niu等[34]在2019年研究发现FTO与乳腺癌有高度相关性,超过70%乳腺癌组织FTO过度表达;且与正常乳腺癌肿瘤组织相比,那些FTO高表达的组织细胞中的m6A甲基化水平降低。之后他们验证了FTO是通过增加促凋亡因子B淋巴细胞瘤-2基因/腺病毒E1B相互作用蛋白3(BCL2/adenovirus E1B 19kDa interacting protein 3,BNIP3)的3'-非翻译区(3'-untranslated region,3'-UTR)去甲基化水平,诱导其降解,从而促进乳腺癌细胞的增殖和转移。RNA-Seq分析确定BNIP3是FTO介导的m6A修饰的下游靶点,在FTO敲低的MDA-MB-231和MCF-7细胞系中均可观察到BNIP3显著上调,说明FTO可以作为乳腺癌的潜在治疗靶标。此外,Xu等[26]研究发现,FTO在人类表皮生长因子受体2(human epidermal growth factor receptor-2,HER2)阳性乳腺癌细胞和组织高表达;且FTO异常表达量与肿瘤大小、细胞核分级和淋巴结转移等显著相关。Kaplan-Meier曲线绘制总生存期与无病生存期表明,高FTO表达导致预后不良以及肿瘤的复发风险增加。在SKBR3和MDA-MB-453细胞中,FTO通过miR-181b-3p通路加速肿瘤细胞的迁移和侵蚀。最新研究发现,衰老中性粒细胞衍生的外泌体piRNA-17560增强了乳腺癌细胞中FTO的表达,FTO的上调通过降低m6A甲基化进一步加强ZEB6转录本的稳定性和表达,导致肿瘤细胞的化学耐药性和上皮-间充质转化(epithelial-mesenchymal transition,EMT)[35]。
除此之外,FTO还与肝癌的发生有关。多项实验证明了FTO可促进肝癌细胞的体外增殖和转移,如Ye等[27]利用siRNA敲低人肝癌细胞系HepG2的FTO基因后,检测到m6A的水平显著上升;CCK-8细胞增殖实验表明下调FTO表达能抑制肝癌细胞的增殖;细胞划痕实验表明,FTO能促进肝癌细胞的转移与侵袭;而在肝内胆管细胞癌(intrahepatic cholangiocarcinoma,ICC)中,FTO表现出抑癌作用。Rong等[36]发现FTO可能通过降低致癌基因(TEAD2和CMTM4)表达和促进抑癌基因(HAO2、NR5A2、CCL19、TCF21、NTRK2和SCML4)表达来抑制ICC的发生。肺腺癌中FTO下调会增加YTHDF1与MYCmRNA的结合,促进MYCmRNA翻译,进而促进肿瘤细胞糖酵解、增殖和肿瘤发生[37]。
在胰腺癌(pancreatic cancer,PC)中,FTO表达水平高于正常胰腺导管上皮细胞,FTO损失导致EMT性状逆转、肿瘤形成受损,并降低癌症干细胞标志物的表达[38]。此外,FTO还可以通过降低m6A-YTHDF1介导的TFPI-1 mRNA的稳定性来促进胰腺癌的发展[39]。在口腔鳞片状细胞系中敲低FTO表达,导致编码真核翻译起始因子γ1基因(eIF4G1)下调,同时增强自噬通量和抑制肿瘤发生[40]。FTO还能通过FTO/miR-576/CDK6通路在膀胱癌中表现出致癌作用,并可能成为膀胱癌的潜在诊断或预后生物标志物[30]。
1.2 m6A去甲基化酶ALKBH5
2013年,Zheng等[6]发现了另一种m6A去甲基化酶ALKBH5,并发现它与生殖细胞的形成相关,在小鼠睾丸中检测到高浓度的ALKBH5表达。敲除小鼠ALKBH5基因后检测到小鼠的精子异常,产生的精子数量减少,小鼠生育能力受损。与FTO相比,ALKBH5对m6A具有更高的特异性。FTO的作用底物较为宽泛,对m6A、m3T、m3U都能进行去甲基化修饰;而目前发现ALKBH5只对m6A有催化活性[22]。FTO对双链DNA、单链DNA和单链RNA在体外都有催化活性,而ALKBH5只特异性催化单链DNA和单链RNA。分析其原因可能是ALKBH5蛋白的氨基酸残基Cys-230和Cys-267之间形成了二硫键,从而阻碍了双链DNA进入ALKBH5的催化结合位点(见图5)[41]。

图5 ALKBH5 Cys-230与Cys-267之间二硫键示意图Figure 5 Diagram of the disulfide bond between ALKBH5 Cys-230 and Cys-267
ALKBH5与FTO一样,通过催化m6A去甲基化,调控m6A影响后续生理过程,如RNA剪切、翻译和降解。ALKBH5的异常表达与许多肿瘤发生相关,如胶质母细胞瘤[42]、乳腺癌[43]、急性髓细胞白血病[44]、肝癌[45]、卵巢癌[46]和非小细胞肺癌[47]等。
2016年,Zhang等[43]报道ALKBH5在乳腺癌的发生发展过程中发挥着关键作用。当乳腺癌细胞暴露在缺氧环境下时会产生缺氧诱导因子(hypoxia inducible factor 1 alpha subunit,HIF-1α)和HIF-2α;而ALKBH5是HIF-1α的直接靶标,从而介导了ALKBH5的过量表达[48]。ALKBH5发挥m6A去甲基化作用,将NANOG基因的3'-UTR上的m6A残基去甲基化,mRNA的稳定性增加,从而引起NANOG高表达。导致乳腺癌细胞在缺氧肿瘤微环境中的富集,促进了体内肿瘤的形成。2017年Zhang等[42]研究发现在胶质母细胞瘤干细胞中ALKBH5高表达,沉默ALKBH5基因能抑制肿瘤的增殖。ALKBH5的异常表达能通过调控FOXM1来增强胶质母细胞瘤的自我更新,并导致肿瘤的产生。转录因子FOXM1与胶质母细胞瘤干细胞的增殖、自我更新等密切相关。
2020年,Shen等[49]研究揭示了ALKBH5可能在AML中发挥促癌作用。他们分析了TCGA以及其他若干AML数据库,发现ALKBH5下调在AML中非常罕见。接下来的一系列机制功能研究揭示了ALKBH5通过调控稳定转录相关酸性卷曲蛋白3基因(transforming acidic coiled-coil-containing protein 3,TACC3)转录产物的mRNA,导致TACC3表达增加(见图6);且TACC3在多种肿瘤中异常表达,在肿瘤的发生和白血病干细胞的自我更新中发挥着重要作用。2020年,Wang等[44]发现ALKBH5是AML肿瘤发生的一个关键调控因子,白血病干细胞染色质的改变通过KDM4C-ALKBH5-AXL信号通路诱导AML发生。ALKBH5的表达是受染色质状态改变的调控,KDM4C能通过增加MYC和PolⅡ的染色质开放性来调控ALKBH5的表达。2023年,Li等[50]发现肌苷三磷酸酶(inosine triphosphatase,ITPA)是ALKBH5的重要功能靶标。在机制上,ALKBH5使ITPA mRNA去甲基化并增加其mRNA稳定性,从而增强ITPA表达。此外,转录因子TCF15在白血病干细胞/起始细胞(leukemia stem/initiating cells,LSC/LIC)中特异性表达,负责ALKBH5在t(8; 21)AML中的表达失调。

图6 ALKBH5促进AML的发生[49]Figure 6 ALKBH5 promotes tumorigenesis in acute myeloid leukemia
Zhu等[46]研究发现ALKBH5通过miR-7和Bcl-2通路抑制上皮性卵巢癌的自噬。他们发现与正常卵巢组织相比,卵巢癌组织中的ALKBH5表达量增加。机制研究证明,ALKBH5可以激活表皮生长因子受体(epidermal growth factor receptor,EGFR)-磷脂酰肌醇3-激酶(phosphatidylinositide 3-kinase,PI3K)-蛋白激酶B(protein kinase B,PKB,又称AKT)-哺乳动物雷帕霉素靶蛋白(mammalian target of Rapamycin,mTOR)信号通路,调控肿瘤细胞的增殖、迁移以及自噬。在淋巴结转移的卵巢癌中,ALKBH5过表达还逆转了ITGB1 mRNA中的m6A修饰,并抑制了YTHDF2蛋白介导的m6A依赖性ITGB1 mRNA降解,导致ITGB1表达增加以及黏着斑激酶(focal adhesion kinase,FAK)和Src原癌基因蛋白的磷酸化,从而增加淋巴结转移[51]。
除了上述促癌情况外,ALKBH5还在多种肿瘤疾病中发挥抑癌作用。如Chen等[45]发现ALKBH5在肝癌中表达下调[45];体外实验证明,ALKBH5能在体外抑制肝癌细胞的增殖和侵蚀能力。含LY6/PLAUR结构域1(LY6/PLAUR domain containing 1,LYPD1)为可能的下游靶点,利用MeRIP-qPCR实验检测发现敲除ALKBH5可以使LYPD1的3'-UTR m6A水平上升,而ALKBH5表达上调可导致该位点m6A水平下降。2023年,Wang等[52]发现ALKBH5通过以m6A依赖性方式下调孕激素和脂联素分子受体4(progestin and adipoQ receptor family member 4,PAQR4)表达来抑制肝癌细胞生长,从而抑制PI3K/AKT通路的激活。Guo等[53]研究发现ALKBH5可以通过m6A-YTHDF2依赖性方式转录激活周期昼夜节律调节器1基因(period circadian regulators 1,PER1)来抑制胰腺癌的发展。Zhang等[54]发现,在胰腺癌中,DDIT4-AS1被确定为ALKBH5的下游靶标之一,ALKBH5介导的m6A修饰导致DDIT4-AS1过表达,DDIT-AS1通过破坏DDIT4的稳定性和激活mTOR途径来增加癌症干性并抑制对吉西他滨的化学敏感性。在结直肠癌中,ALKBH5下调与结直肠癌患者预后不良密切相关,敲低ALKBH5增强了体外LOVO细胞的增殖、迁移和侵袭,ALKBH5通过降低PHF20 mRNA甲基化来抑制结直肠癌进展。ALKBH5介导的PHF20 mRNA的m6A修饰可以作为干预和治疗结直肠癌的有效策略[55]。
1.3 DNA修复蛋白ALKBH3
2005年Konishi等[56]在前列腺中分离得到一种高表达的蛋白,最初被命名为前列腺癌抗原-1(prostate cancer antigen-1,PCA-1)。随后的研究发现它是Alkb的同源蛋白,故称为ALKBH3[57]。ALKBH3是一种DNA修复蛋白,可以通过氧化脱烷基对烷基化损伤的DNA进行修复,对单链DNA和单链RNA有着高选择性,能去甲基化m1A和m3C。近期研究还发现其对tRNA具有去甲基化作用[7]。ALKBH3在多种恶性肿瘤中存在高表达,如非小细胞肺癌[58]、胰腺癌[59]和前列腺癌[56]等,并且与肿瘤的不良预后显著相关。因此ALKBH3可能是某些肿瘤治疗的潜在靶点。
Koike等[57]探究了ALKBH3与前列腺癌的相关性,并发现前列腺癌中ALKBH3高表达,但在良性前列腺增生和正常前列腺上皮细胞中正常表达。通过siRNA转染实验敲低非激素依赖的前列腺癌细胞株中的ALKBH3,抑制了细胞的生长以及诱导细胞凋亡。实体瘤小鼠实验也证明,抑制ALKBH3能显著抑制肿瘤的生长,推测是通过PI3K-AKT信号通路调节;但具体的RNA去甲基化与AKT活性之间的关系尚不明确。针对ALKBH3开发抑制剂可能是治疗去势耐药前列腺癌(castrate resistant prostate cancer,CRPC)的有效方法。2012年,Yamato等[59]发现ALKBH3与胰腺癌发生有关。在大多数人类胰腺癌组织中ALKBH3高表达,在非癌组织中未见表达。利用siRNA转染敲低胰腺癌细胞系PANC-1和MIAPaCa-2上的ALKBH3基因,观测到细胞凋亡和抗血管生成。随后,他们又研究了ALKBH3与血管内皮生长因子(vascular endothelial growth factor,VEGF)的关系,推测VEGF与胰腺癌的发生密切相关。结果显示随着细胞内ALKBH3的下调,VEGF的表达显著降低,提示ALKBH3与血管生成相关,可能是VEGF的上游通路。
2011年,Tasaki等[58]通过siRNA转染实验发现,在体内或体外抑制ALKBH3的表达均能诱导非小细胞肺癌(non-small cell lung carcinoma,NSCLC)的衰老并抑制其生长。他们还发现,随着ALKBH3被敲低,p21和p27的水平也下降,推测抑制ALKBH3可以下调p21和p27的表达,从而抑制肿瘤生长。p21和p27均是细胞周期蛋白依赖性激酶抑制剂家族中的重要成员,既与肿瘤抑制作用密切相关,又能通过抑制周期素依赖激酶(cyclindependent kinases,CDKs)复合物活性,协调细胞周期、DNA复制与修复之间的关系。此外,2017年Kogaki等[60]发现ALKBH3与TP53基因有关。在NSCLC中,降低ALKBH3蛋白的表达会导致细胞周期阻滞或者凋亡,这取决于TP53基因的状态。携带野生型TP53基因的NSCLC细胞阻滞停在G2/M期;而携带突变TP53基因的NSCLC细胞则发生细胞凋亡。
除了前列腺癌、胰腺癌和非小细胞肺癌外,有研究指出ALKBH3能通过去甲基化调控巨噬细胞集落刺激因子-1(colony stimulating factor-1,CSF-1)mRNA中的m1A,延长了CSF-1的半衰期,调控卵巢癌和乳腺癌的发生[61]。然而也有相关报道,ALKBH3在乳腺癌中表达量不高,并且检测到大多数ALKBH3的启动子CpG区域发生甲基化修饰,导致ALKBH3表达降低,DNA m3C烷基化损伤积累[62]。有研究表明乳腺癌的发生与DNA修复能力缺陷相关,而ALKBH3正是一种DNA修复蛋白,因此ALKBH3下调可能与乳腺癌发生有关。
2 m6A去甲基化酶小分子抑制剂
2.1 FTO小分子抑制剂
FTO作为m6A的去甲基化酶,发挥RNA去甲基化作用需要Fe2+和2-OG辅助。因此其小分子抑制剂主要分为3种类型:金属离子螯合抑制剂、2-OG类似物和m6A竞争性抑制剂。前2类抑制剂选择性不强,开发价值有限。本节主要针对m6A竞争性抑制剂进行综述。
2.1.1 大黄酸Chen等[16]在2012年报道了首个FTO小分子抑制剂。他们通过高通量虚拟筛选以及后续的结构优化,发现了结构新颖的天然产物大黄酸(1)(见图7)。随后利用等温滴定量热法(isothermal titration calorimetry,ITC)、蛋白热漂移实验以及CD圆二色谱等方法验证了大黄酸与FTO蛋白结合能力,利用高效液相色谱法(high performance liquid chromatography,HPLC)测得大黄酸对FTO具有一定的抑制作用,IC50为30 μmol · L-1。为进一步验证大黄酸对FTO的结合模式,他们测试了大黄酸在不同金属离子浓度条件下对FTO的抑制活性。因其活性不受金属离子浓度的影响,故推测大黄酸是m6A的底物竞争性抑制剂。随后测试了大黄酸在细胞水平上对FTO的抑制能力,在BE(2)-C细胞中给药24 h后监测到明显的m6A水平上升,证明大黄酸可以调节细胞内的m6A水平。

图7 化合物FB23-2(4)的优化过程Figure 7 Structural optimization of FB23-2(Compound 4)
2.1.2 甲氯灭酸及其衍生物Huang等[17]继而在2015年报道了一种FTO的选择性小分子抑制剂。通过高通量荧光偏振筛选方法,得到对FTO与ALKBH5具有选择性的抑制剂——甲氯灭酸(meclofenamic acid,MA,2)。通过DNA PAGE实验验证了MA在酶水平上对FTO有抑制活性(IC50为30 μmol · L-1),而对ALKBH5没有活性。进一步利用MA与FTO晶体复合物的信息确定了FTO核酸识别序列(nucleotied recognition lid,NRL)中的一段loop区与MA有疏水相互作用(见图8);而ALKBH5中缺少这段loop区,阐明了MA对FTO的选择性抑制原因。随后2019年该课题组基于对MA进行的一系列结构改造,得到了活性更好的化合物FB23(3,IC50为60 nmol · L-1)(见图7和8)[18]。尽管FB23的靶标活性很高,但由于其理化性质不佳导致细胞活性较差(在NB4细胞系中抑制活性为44.8 μmol · L-1,在MONOMAC6中抑制活性为23.6 μmol · L-1),为改善FB23的溶解性和透膜性,将FB23的羧基用羟胺缩合封闭得到了化合物FB23-2(4),FB23-2在NB4和MONOMAC6细胞系的抗细胞增殖活性得到了很大提升,分别为0.8和1.5 μmol · L-1。FB23-2在体外可以显著地抑制AML细胞增殖,促进细胞分化与凋亡,而对敲除FTO的AML细胞未显示出明显的抗增殖活性。进一步的研究验证了FB23-2是通过抑制m6A去甲基化酶FTO的活性,导致AML细胞中m6A水平上升,并抑制了下游通路MYC和CEBPA基因的表达。

图8 MA及FB23与FTO结合模式Figure 8 Combination mode of MA or FB23 with FTO
2.1.3 CS1和CS2Su等[63]在2020年也报道了2个FTO小分子抑制剂,其在细胞上对FTO的抑制活性优于FB23-2,达到了纳摩尔水平。通过基于结构的虚拟筛选策略以及后续细胞抗增殖实验和荧光偏振实验,从NCI DTP数据库中筛到2个在酶靶标水平对FTO有抑制活性的小分子CS1(5)和CS2(6)。CS1是一种含有蒽环结构的化合物,从20世纪80年代开始便被作为多种癌症的临床药物使用。CS2是一种二氢乳清酸脱氢酶抑制剂,能阻断嘧啶的生物合成。通过FTO蛋白与化合物的核磁共振分析(nuclear magnetic resonance,NMR)观察到剂量依赖的信号衰减,证明了CS1、CS2确实能与FTO蛋白结合。随后的细胞抗增殖实验表明,在FTO高表达的AML细胞系中该化合物表现出很好的抑制活性,而在FTO低表达的AML细胞中活性变弱。对FTO高表达的细胞株进行FTO基因的敲除后,细胞对CS1和CS2的敏感度降低,这说明CS1和CS2对细胞的抑制与FTO的表达有关,间接说明了化合物通过抑制FTO来调控m6A水平。之后的小鼠AML动物模型实验也证明CS2可以改善白血病状况,提高小鼠的整体存活率。
2.1.4 18077和18097Xie等[64]于2022年报道了2种FTO小分子抑制剂。通过虚拟筛选及结构优化,发现了化合物18077(7)和18097(8)可以选择性地抑制FTO的去甲基化酶活性。18097通过结合到FTO的活性部位,从而抑制细胞周期过程和癌症细胞的迁移和侵袭。此外,18097对人乳腺癌细胞的表观转录组进行了重新编程,尤其是与P53通路相关的基因,18097增加了细胞因子信号传导抑制因子1基因(SOCS1)的mRNA水平,其募集IGF2BP1以增加SOCS1的mRNA稳定性,随后激活P53信号通路。此外,18097通过下调过氧化物酶体增殖物激活受体γ(peroxisome proliferatoractivated receptor γ,PPARγ)、CCAAT/增强子结合蛋白α(C/EBPα)和C/EBPβ来抑制细胞脂肪生成。动物研究证实,18097可以显著抑制乳腺癌细胞的体内生长和肺部定植。
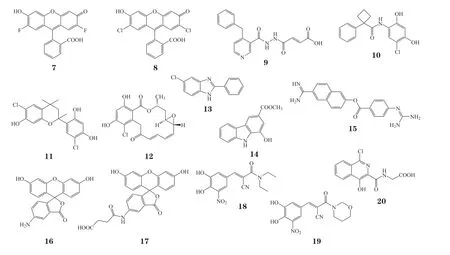
2.1.5 其他抑制剂除了上述抑制剂外,还有一些课题组报道了部分FTO小分子抑制剂。如Toh等[22]在2015年报道的系列化合物,其活性最好的化合物9对FTO的酶抑制活性为0.81 μmol · L-1。
He等[65]发现化合物N-CDPCB(10),酶活性IC50为4.95 μmol · L-1。2016年发现CHTB(11),通过蛋白-晶体复合物信息发现其与N-CDPCB的结合模式类似,IC50为39.24 μmol · L-1。2018年报道的根赤壳菌素(Radicicol,12),IC50为16.04 μmol · L-1,并通过ITC手段确证其可与蛋白在体外结合[66]。2019年探究了一类苯并咪唑类化合物2位氯取代对FTO活性的影响,该类化合物13的IC50为24.65 μmol · L-1[67]。同年发现了一种咔唑类天然产物Clausine E(14),IC50为27.8 μmol · L-1[68]。2019年该课题组通过老药新用方法,发现萘莫司他苯磺酸盐(15),IC50为13.77 μmol · L-1[69]。
Wang等[70]发现荧光素衍生物FL1(16)(FTO IC50= 6.55 μmol · L-1)和FL2(17)(FTO IC50= 1.72 μmol · L-1)作为双功能分子,可以同时抑制和标记FTO蛋白,选择性抑制细胞内外的FTO去甲基化。通过晶体学阐明该荧光抑制剂的构效关系,并为FTO去甲基化抑制剂的结构优化提供方向。此外,该策略还实现了FTO的下游生化分析,如活细胞中FTO蛋白的可视化和FTO富集。该荧光抑制剂可作为潜在的工具分子以研究FTO在m6A介导的表观遗传过程中发挥的作用。
2019年,Peng等[71]通过虚拟筛选和分子对接等手段筛选已上市药物数据库,得到FTO抑制剂恩他卡朋(Entacapone,18),IC50为3.5 μmol · L-1。通过对该化合物的构效关系研究得到活性化合物19,其IC50为0.7 μmol · L-1。借助蛋白-化合物晶体复合物明确了恩他卡朋与FTO的结合模式。
McMurray等[72]在2015年报道了IOX3(20),是一种脯氨酰羟化酶(prolyl hydroxylase domain protein,PHD2)抑制剂;但之后的研究表明它对多种2-OG加氧酶例如FTO、PHD2都有抑制活性,能非共价竞争结合催化位点,其对FTO的IC50为2.76 μmol · L-1。
2.2 ALKBH3小分子抑制剂
2014年,Nakao等[73]报道了一系列ALKBH3抑制剂。他们通过虚拟筛选市售化合物库中的17 000个小分子,得到苗头化合物21,后续的结构经优化得到一系列苯并咪唑类化合物。其中HUHS015(22)为活性最好的化合物,在体外实验和小鼠移植瘤模型中均有良好的抑制率。为了提高溶解度,该课题组研究了HUHS015不同种类的盐,并最终采用钠盐(23)开展后续药代动力学实验[74]。遗憾的是,HUHS015在大鼠体内不稳定,容易被肝代谢失活。用大鼠肝均浆在15℃处理HUHS015 10 min后仅剩42%,37℃条件下完全被代谢[75]。为改善化合物溶解度以及易被肝代谢的特点,他们进行了新一轮结构优化,最终得到化合物HUHS119(24)。经口给药后能在血清中保持一定浓度,并且在酶水平(IC50= 2.9 μmol · L-1)和细胞水平(10 μmol · L-1时抑制率为81%)上有着较好抑制活性(见图9)。

图9 化合物HUHS119(24)的优化过程Figure 9 Structural optimization of HUHS119(Compound 24)
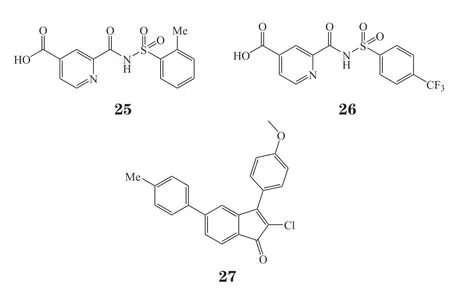
2018年Das等[76]利用动态组合化学的手段筛选得到苗头化合物,将其中相对不稳定的羰基酰腙基团结构替换成相对稳定的羰基磺酰胺结构,得到一系列ALKBH3和FTO的选择性抑制剂。利用蛋白热漂移实验和HPLC验证化合物对ALKBH蛋白家族的结合活性及酶水平活性,发现了ALKBH3选择性抑制剂化合物25和FTO选择性抑制剂化合物26。利用分子对接软件分析,发现可能是由于化合物26的4-三氟甲基苯基的空间构象原因,与Tyr143产生位阻,无法伸入ALKBH3结合口袋。但这无法解释化合物25对ALKBH3的选择性,还需要后续共晶复合物研究辅助分析。同年,Nigam等[77]报道了一系列二氢茚酮类衍生物,对ALKBH3有着中等抑制活性。后续利用ITC结合实验、HPLC酶活实验等验证了化合物27对蛋白的靶标活性,并进行了细胞抗增殖实验。在非小细胞肺癌细胞系A549中测得EC50约为18.7 μmol · L-1,还需进行后续的结构改造优化工作。
2.3 ALKBH5小分子抑制剂
同样作为m6A的RNA去甲基化酶,对FTO的同源蛋白ALKBH5的小分子抑制剂相关研究相对较少,还处于起步阶段。目前已经报道的几种抑制剂多数属于泛抑制剂,包括N-草酰乙二酰甘氨酸(N-oxalylglycine,NOG)和2,4-吡啶二羧酸(pyridine 2,4-dicarboxylate,2,4-PDCA)等;或是在其他同源蛋白抑制剂研究中发现的非选择性抑制剂,这些泛抑制剂普遍对ALKBH5抑制活性较低。如Chen等[16]在2012年报道的首个FTO小分子抑制剂大黄酸,测试其对FTO同源蛋白的抑制活性,发现大黄酸对ALKBH2、ALKBH3[78]和ALKBH5都有微弱的活性。Das等[76]在2018年报道的FTO和ALKBH3选择性抑制剂中,也有若干化合物对ALKBH5有微弱活性。最近有研究报道了基于荧光共振转移法筛选得到对FTO抑制的天然产物[79]。其中一些天然产物对ALKBH5也有泛抑制作用,如大黄素、茜素红、白花丹素等。
前述化合物IOX3(16)是首个报道的可与ALKBH5产生共价作用的抑制剂[21],通过共晶衍射手段,IOX3能特异性地与ALKBH5结构域中的Cys200以二硫键形式结合。IOX3最初作为PHD抑制剂被开发;但之后的研究发现其对2-OG加氧酶,如FTO、PHD2等均存在抑制活性,能非共价竞争结合于催化位点,对于ALKBH5而言则是通过芳香亲核取代反应共价结合在氨基酸残基Cys200上(见图10)。

图10 IOX3与ALKBH5结合模式Figure 10 Combination mode of IOX3 with ALKBH5
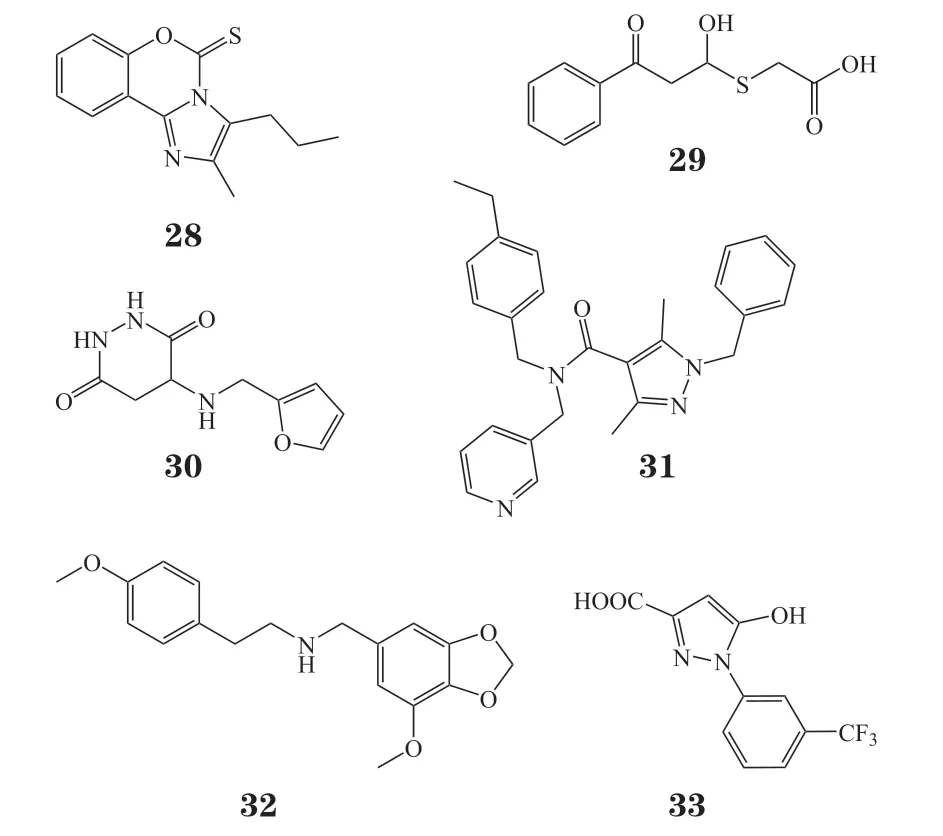
Malacrida等[80]报道了ALKBH5抑制剂MV1035(28)。他们通过SPILLO-PBSS软件反向筛选MV1035可能作用的靶点,并通过斑点印记实验、细胞抗增殖实验进行验证。在U87-MG细胞系中MV1035的IC50为(2.48±0.65)μmol · L-1;但该研究并未进行靶标层面的结合实验,只有斑点印记实验辅助进行了定性验证。除此之外,Li等[81]也报道了一个ALKBH5抑制剂ALK-04,并进行了小鼠体内抗肿瘤实验,但其结构并未公开。
Selberg等[82]报道了2种ALKBH5抑制剂(化合物29和30),它们可以在低微摩尔范围内抑制3种白血病细胞系HL-60、CCRF-CEM和K562的增殖。Takahashi 等[83]发现新的小分子抑制剂Ena15(31)和Ena21(32),能够抑制ALKBH5的去甲基化活性,而对FTO没有抑制活性,并测试了化合物对人多形性胶质母细胞瘤(glioblastoma multiforme,GBM)细胞抗增殖活性。
2022年,Fang等[84]报道了一类新的含有1-芳基-1H-吡唑骨架的ALKBH5抑制剂。通过基于荧光偏振(fluorescence polarization,FP)的筛选、结构优化和构效关系分析,获得了活性最佳的化合物20m(33)(FP IC50= 21 nmol · L-1)。相对于FTO以及其他ALKB亚家族成员,化合物20m对ALKBH5表现出高选择性。细胞热位移分析(cellular thermal shift assay,CETSA)表明,20m可以有效稳定HepG2细胞中的ALKBH5。斑点印迹分析表明,20m可以提高细胞中的m6A水平。
3 结语与展望
作为中心法则的关键中间环节,RNA将DNA与蛋白质紧密联系。RNA的修饰与后续翻译、RNA降解等过程关系密切。目前,RNA修饰已经成为表观遗传学研究的热门领域。细胞内的RNA中存在着100多种不同的化学修饰,其中甲基化修饰包括m6A、m5C、m1A 等。而最常见最保守的RNA甲基化修饰是m6A,近几年有大量研究揭示了m6A与包括肿瘤、病毒感染等疾病的相关性。
哺乳动物细胞中的m6A修饰具有可逆性。m6A可被m6A阅读蛋白识别,其甲基化和去甲基化过程分别由m6A甲基转移酶和m6A去甲基化酶参与调控。m6A的甲基化动态修饰在多种肿瘤疾病中发挥重要作用。靶向m6A去甲基化酶设计小分子抑制剂来调控m6A动态过程已经成为一种潜在的治疗癌症的新策略。目前基于FTO的相关肿瘤通路、小分子抑制剂研究的较多,已有高活性和高选择性的小分子抑制剂报道,如Huang等[17-18]报道的MA、FB23和FB23-2,Su等[63]报道的CS1和CS2等。针对ALKBH5,目前的小分子抑制剂的研究尚处于起步阶段,还有待进行更多、更深入的生物学和化学研究。ALKBH3最近也逐渐受到人们的关注,小分子抑制剂的报道呈上升趋势,但后续化学生物学相关研究依然较少,并且尚未有更多的文章证据表明ALKBH3针对RNA上m6A位点的修饰。目前报道的m6A去甲基化酶小分子抑制剂大多为2-OG的类似物,如IOX3、大黄酸等。这些化合物的主要问题是选择性不高,对其他2-OG依赖的酶也有抑制作用。针对底物m6A结合区域设计选择性小分子抑制剂是未来的发展趋势之一,如FB-23针对FTO和ALKBH5的一段疏水loop区实现对FTO蛋白的选择性抑制。开发更多结构类型的FTO选择性抑制剂以及对FTO抑制后的生物学效应研究依然是后续研发的重点。而如何针对ALKBH家族亚型的细微差异实现ALKBH5、ALKBH3等蛋白之间的选择性将是其他m6A去甲基化酶小分子抑制剂研发的重点和难点。
作为目前RNA修饰研究热点领域,大量研究发现m6A与肿瘤等疾病的相关性[25-28];但依然有很多问题待研究解决。如导致肿瘤疾病的具体分子机制,m6A修饰的下游信号通路以及相关RNA如何做到特定的甲基化修饰等。针对m6A去甲基化酶报道的小分子抑制剂有望成为关键的工具分子,帮助研究人员进一步阐明m6A的相关生物学机制。希望在国内外研究者的推动下,未来可以针对特定肿瘤,开发出安全有效的m6A去甲基化酶抑制剂来调控m6A水平,进而干扰肿瘤的发生和发展,推动肿瘤疾病的治疗。
