表面增强拉曼散射技术在快速检测预包装干米粉中乙二胺四乙酸二钠的应用
2023-10-21孔红星陈绮莹蒙海森陈瑞珏
孔红星,陈绮莹,蒙海森,陈瑞珏,冯 军,程 昊,*
(1.广西糖资源绿色加工重点实验室,广西柳州螺蛳粉工程技术研究中心,广西科技大学生物与化学工程学院,广西 柳州 545006;2.蔗糖产业省部共建协同创新中心,广西 南宁 530004;3.广西科技大学医学部,广西 柳州 545000)
乙二胺四乙酸二钠(ethylenediaminetetraacetic acid disodium salt,EDTA-2Na)是一种广泛使用的食品添加剂。EDTA-2Na分子中含有强配位能力的氨基和羧基,能与食品中微量的金属离子形成稳定的络合物,有效地防止由于金属离子作用而导致的食品褪色、浑浊、变质以及VC氧化[1]。EDTA-2Na因具有较强的络合能力,对食品有护色、稳定以及抗氧化的作用,能有效地保护食品中的营养成分,而常作为食品添加剂添加到食品中[2]。根据GB 2760—2014《食品添加剂使用标准》规定,EDTA-2Na允许在果脯、蔬菜罐头和复合调味品中作为稳定剂、凝固剂、抗氧化剂和防腐剂使用[3]。
随着食品工业的发展,预包装柳州螺蛳粉因其鲜香酸辣爽的特性,被广大消费者所熟知。干制米粉作为预包装柳州螺蛳粉的主食,因其顺滑且有嚼劲的口感而逐渐受到人们的青睐,但GB 2760—2014中允许大米制品使用的食品添加剂极少[3-4]。然而为了提高预包装干米粉的口感和延长保质期,个别商家会在干米粉中超量添加食品添加剂——EDTA-2Na,但米粉并不在允许添加EDTA-2Na作为食品添加剂的食品范围内[5-7]。EDTA-2Na的过量使用,会对上呼吸道、眼睛、黏膜产生影响,甚至会引发呕吐、腹泻和急性腹痛等症状,对人体健康造成严重影响[8]。为了更好地保护预包装干米粉市场,开发一种可以快速、灵敏地检测预包装干米粉中EDTA-2Na的方法很有必要。
目前,检测食品中EDTA-2Na的方法有气相色谱-质谱法[9]、化学发光[10-11]、荧光[12]、比色法[13]和高效液相色谱(high performance liquid chromatography,HPLC)等[14-15]。这些方法尽管检测结果精确,但样品前处理繁琐、检测过程复杂,对EDTA-2Na的检出限较高。相关研究[5,7,15]表明,利用HPLC法检测小麦粉或面粉中EDTA-2Na的检出限为2.00~11.15 mg/kg,难以满足微量添加EDTA-2Na的预包装干米粉的检测。表面增强拉曼散射(surface enhanced Raman scattering,SERS)是一种基于激光光子非弹性散射的振动光谱,由于电磁增强和化学增强使SERS基底与待测物分子结合时会发生表面等离子共振相互作用,而引起拉曼散射增强的现象。SERS光谱是一种可以对痕量分子进行快速、灵敏分析的检测技术,对待测物具有“指纹图谱”的优势,可准确鉴定痕量物质[16-17]。与出入境检验检疫行业标准(SN/T 3855—2014《出口食品中乙二胺四乙酸二钠的测定》)使用的液相色谱测定法相比,SERS检测简化了前处理过程,无需采用三氯甲烷净化米粉中的大分子化合物,大大缩短了检测时间。除此之外,SERS能提供样品的分子结构信息,因具有灵敏度高、检测过程快速、便捷等优点,而成为一种可灵敏检测市售预包装干米粉中EDTA-2Na添加情况的技术[18]。
磁性银纳米颗粒具有良好的纳米核壳结构,磁性核心有利于其富集目标分子并实现快速分离,亲水性、生物相容性良好的SiO2层可增强Fe3O4纳米粒子的稳定性。除此之外,Fe3O4上密集、均匀沉积的银纳米颗粒(Ag nanoparticles,AgNPs)有利于提高SERS强度,这是由于磁性核心可以调节银纳米粒子的聚集状态,使其产生更丰富的热点,热点内的电磁耦合作用导致电磁场强度增强,从而达到更好的SERS增强效果[19-22]。
本研究制备了具有良好SERS活性的Fe3O4@SiO2@AgNPs,并以其为拉曼基底检测预包装干米粉中EDTA-2Na的含量。目前,利用SERS技术检测预包装干米粉中EDTA-2Na含量的研究鲜见报道,因此,本实验建立一种基于SERS的快速、灵敏检测预包装干米粉中EDTA-2Na的方法,以满足简单、迅速、准确地检测预包装干米粉中痕量添加剂——EDTA-2Na的需要。
1 材料与方法
1.1 材料与试剂
螺蛳粉 东莞市一家人食品有限公司、广西螺霸王食品有限公司、柳州市好欢螺食品有限公司。
EDTA-2Na(分析纯)、硝酸银(分析纯)、罗丹明6G(rhodamine 6G,R6G,生物染色剂)、微晶纤维素((C6H10O5)n,柱层析) 国药集团化学试剂有限公司;乙二醇(分析纯) 成都市科隆化学品有限公司;六水合三氯化铁(分析纯) 台山市粤侨塑料有限公司;醋酸钠(分析纯) 广州市金华大化学试剂有限公司;正硅酸乙酯(tetraethyl orthosilicate,TEOS)、三水合乙酸铅、五水合硫酸铜、氯化钠(均为分析纯) 西陇化工股份有限公司;氨水(分析纯) 广东光华科技股份有限公司;聚乙烯吡咯烷酮K30(polyvinyl pyrrolidone K30,PVP K30,mw=58 000) 上海麦克林生化科技有限公司;可溶性淀粉((C6H10O5)n,分析纯)天津市登峰化学试剂厂;氯化钾、抗坏血酸(均为分析纯) 广东光华科技股份有限公司。
1.2 仪器与设备
XploRA PLUS激光共聚焦拉曼光谱仪 日本Horiba公司;S-4800冷场发射扫描电子显微镜 日本日立公司;JEOL JEM 2100透射电子显微镜 日本电子株式会社;50 MAX EDS能谱仪 英国牛津仪器公司;D8A A25 X射线衍射(X-ray diffraction,XRD)仪、INVENIO R傅里叶变换红外光谱仪 德国布鲁克公司。
1.3 方法
1.3.1 材料制备与表征
1.3.1.1 材料制备
Fe3O4@SiO2@AgNPs的制备过程参考文献[23-25],具体流程如图1所示。操作要点如下:

图1 Fe3O4@SiO2@AgNPs的制备流程图Fig.1 Schematic diagram of fabrication of Fe3O4@SiO2@AgNPs
1)合成Fe3O4:取1.35 g FeCl3·6H2O与2.7 g醋酸钠混合于40 mL乙二醇中,磁力搅拌至所有反应物完全溶解,上述混合物在高压反应釜内190 ℃条件下持续反应10 h,冷却至室温,在外加磁场的作用下,用去离子水和无水乙醇交替洗涤产物3~4 次,真空干燥1 h后得到Fe3O4磁性纳米粒子。
2)Fe3O4@SiO2的制备:取0.2 g Fe3O4磁性纳米粒子超声分散于60 mL无水乙醇中,在持续超声条件下加入4 mL去离子水,0.2 mL TEOS和1 mL 25%的氨水。继续超声1 h后,每隔1 h加入0.2 mL TEOS,共加4 次。待反应完全后,在外加磁场的作用下,清洗并干燥反应产物,步骤同上,得到Fe3O4@SiO2纳米粒子。
3)Fe3O4@SiO2@AgNPs的制备:将0.5 g PVP溶于26 mL无水乙醇中,超声30 min。精密称取2 g硝酸银溶解在12 mL去离子水中,随后滴加25%氨水,一边滴加一边振荡,直到固体刚好消失停止滴加氨水,并将其加入到含有PVP的无水乙醇溶液中。在上述混合溶液中加入0.2 g Fe3O4@SiO2超声10 min,在高压反应釜内120 ℃条件下持续反应4 h。待反应完全后,清洗并干燥反应产物,步骤同上,烘干即得Fe3O4@SiO2@AgNPs。
1.3.1.2 材料表征
采用透射电子显微镜对合成材料的形貌结构进行观察,并使用EDS能谱仪对材料的元素分布进行表征。
利用XRD仪采集材料在扫描角度(2θ)为10°~80°的X射线衍射谱,工作电压与工作电流分别为40 kV与40 mA。
将干燥的材料与KBr混合研磨后压片,使用傅里叶变换红外光谱仪分析材料的结构。
1.3.2 Fe3O4@SiO2@AgNPs的SERS活性
称取4.79 mg R6G粉末加入去离子水定容到10 mL容量瓶中,而后将母液稀释到10-4~10-8mol/L。取100 μL Fe3O4@SiO2@AgNPs(1 mg/mL)与100 μL不同浓度的R6G溶液混合,在室温下超声30 min,取10 μL混合液滴在干净的载玻片上,放置在25 ℃、相对湿度22%的环境中,直至溶液水分自然挥发后采集拉曼信号,每个样品重复3 次取平均值。使用过程中拉曼光谱的各项参数为:激发波长638 nm,10 倍物镜,积分时间10 s,功率为1.6 mW,累积次数2 次。
1.3.3 EDTA-2Na的SERS检测
1.3.3.1 EDTA-2Na标准溶液的制备及SERS检测
称取0.1 g EDTA-2Na配成质量浓度为10.00 mg/mL的标准储备溶液。测量前将EDTA-2Na标准溶液稀释成质量浓度分别为0.01、0.1、1、10、100、1 000 μg/mL的EDTA-2Na标准工作溶液。
取100 μL Fe3O4@SiO2@AgNPs(1 mg/mL)与100 μL不同质量浓度的EDTA-2Na溶液混合,在室温下超声20 min。取10 μL混合溶液滴在干净的载玻片上,放置在25 ℃、相对湿度22%的环境中,直至溶液水分自然挥发后进行拉曼检测,每个样品重复3 次取平均值。使用过程中拉曼光谱的各项参数与1.3.2节相同。
1.3.3.2 样品溶液的SERS检测
制备预包装螺蛳粉干米粉中的EDTA-2Na待测液:参考GB 5009.278—2016《食品中乙二胺四乙酸盐的测定》配制[25]。
取预包装螺蛳粉干米粉500 g用捣碎机磨成粉末状,称取5 g粉末(精确至0.01 g),分成5 份,分别加入10 mL离心管中,每管加入5 mL去离子水,充分涡旋后,超声20 min。高速离心后将上清液集中到50 mL容量瓶中。在每管沉淀物中加入去离子水重复提取,合并提取液于50 mL容量瓶中,用去离子水定容到50 mL,经0.45 μm滤膜过滤制成干米粉样品溶液。在干米粉样品溶液中添加0.20~1.00 mg/kg范围内的低、中、高三个浓度的EDTA-2Na,取100 μL不同浓度的待测液与Fe3O4@SiO2@AgNPs超声混合20 min,根据1.3.3.1节方法干燥后进行SERS检测,拉曼光谱的参数与1.3.2节相同,每个样品重复3 次取平均值。
1.4 数据统计与分析
实验结果运用Origin软件进行绘图并进行线性拟合,SERS增强因子按下式计算:
式中:EF为增强因子;ISERS为在Fe3O4@SiO2@AgNPs基底上检测的1×10-8mol/L(CSERS)R6G在1 652 cm-1拉曼位移处的强度;CRaman为能产生普通拉曼信号的R6G浓度(1 mol/L);IRaman为其在1 652 cm-1特征峰的拉曼强度。
2 结果与分析
2.1 Fe3O4@SiO2@AgNPs的表征
2.1.1 透射电子显微镜及X射线能谱(energy dispersive spectrometer,EDS)分析
如图2所示,Fe3O4@SiO2@AgNPs是通过在Fe3O4表面包覆SiO2层后沉积AgNPs得到的。制备得到的Fe3O4(图2a)分散良好,尺寸大小均匀,直径约为490 nm。Fe3O4的EDS元素分析图(图2d)表明Fe3O4纳米颗粒只有Fe和O元素。Fe3O4包覆SiO2层后,形成了典型的核壳式结构,如图2b所示,Fe3O4表面包覆着薄而透明的SiO2层,其厚度约为40~50 nm,Fe3O4@SiO2直径约为600 nm。Fe3O4@SiO2的EDS元素分析图(图2e)表明Fe3O4@SiO2纳米颗粒含有Fe、O和Si元素,证明Fe3O4成功包覆了SiO2层。如图2c所示,合成的Fe3O4@SiO2@AgNPs表面粗糙,大小较为均匀,直径约为660 nm,直径约50 nm的AgNPs均匀地铺满在Fe3O4@SiO2表面。其EDS元素分析图(图2f)表明,制备的纳米复合材料只存在Fe、Ag、Si和O元素,没有存在其他元素表明制造的纳米粒子纯度高。
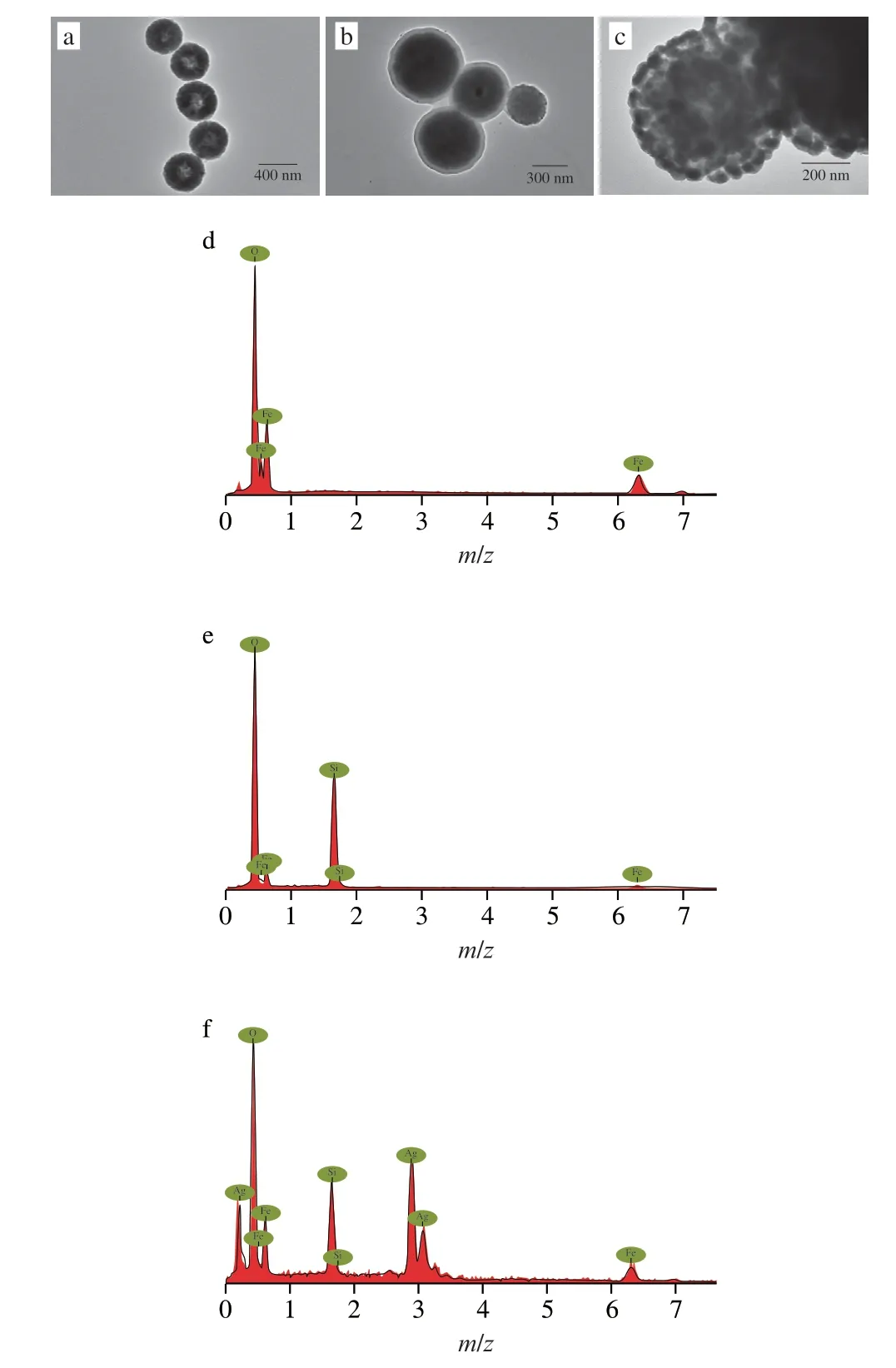
图2 Fe3O4(a)、Fe3O4@SiO2(b)和Fe3O4@SiO2@AgNPs(c)的透射电子显微镜图及Fe3O4(d)、Fe3O4@SiO2(e)和Fe3O4@SiO2@AgNPs(f)的EDS分析图Fig.2 TEM images of Fe3O4 (a), Fe3O4@SiO2 (b) and Fe3O4@SiO2@AgNPs (c) and energy dispersive spectra of Fe3O4 (d), Fe3O4@SiO2 (e) and Fe3O4@SiO2@AgNPs (f)
2.1.2 XRD及傅里叶变换红外光谱图分析
分别对Fe3O4、Fe3O4@SiO2,Fe3O4@SiO2@AgNPs采用XRD进行表征,结果如图3a所示。Fe3O4磁性纳米粒子分别在2θ为18.31°、30°、37°、43°、53.4°、56.9°和62.5°处出现衍射峰,分别对应于尖晶石Fe3O4的(111)、(220)、(311)、(400)、(422)、(511)和(440)晶格平面,符合Fe3O4标准反射(JCPDS卡号75-1609)[27]。由于覆盖在Fe3O4上的SiO2是无定形的,在XRD上不会出峰,因此Fe3O4@SiO2的XRD曲线衍射峰与Fe3O4几乎完全相同。与Fe3O4和Fe3O4@SiO2的XRD曲线相比,Fe3O4@SiO2@AgNPs在2θ为38.3°、44.3°、64.5°和78°处出现4 个额外的峰,与标准银衍射峰(JCPDS No.4-0783)比较,分别对应于面心立方Ag的(111)、(200)、(220)和(311)晶格平面(标有符号*)[28],证明银纳米粒子已经成功生长在磁性颗粒的表面。同时,Fe3O4的所有衍射峰在包裹了SiO2层以及沉积了银纳米粒子后仍被保留,证明磁性Fe3O4颗粒结构未被破坏。同时,没有出现归属不明的峰,说明制作过程并未引入杂质。
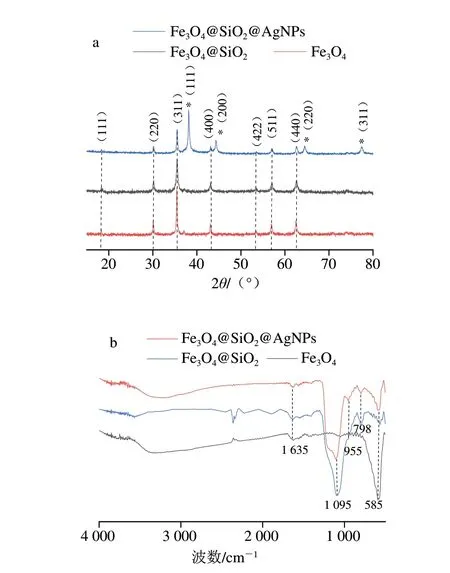
图3 Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@AgNPs的XRD图谱(a)及傅里叶变换红外光谱图(b)Fig.3 XRD spectra (a)and FT-IR spectra (b) of Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@AgNPs
为了进一步确认材料的组成和结构,测量了傅里叶变换红外光谱。如图3b所示,585 cm-1处吸收峰为Fe3O4微球的典型带,这与Fe—O的伸缩振动有关[29]。1 635 cm-l处的峰是水的H—O—H弯曲振动峰。覆盖二氧化硅层后,Fe3O4@SiO2微球显示出以1 095、955 cm-1和798 cm-1为中心的新谱带。1 095、798 cm-1和955 cm-1处的吸收峰分别是Si—O—Si的不对称伸缩振动和对称伸缩振动吸收峰以及Si—OH的弯曲振动吸收峰[30],表明SiO2成功覆盖在Fe3O4微球的表面上。由于AgNPs在红外区域没有吸收[31],因此Fe3O4@SiO2@AgNPs的红外光谱图与Fe3O4@SiO2微球几乎相同。
2.2 Fe3O4@SiO2@AgNPs的拉曼光谱分析
2.2.1 Fe3O4@SiO2@AgNPs对R6G的灵敏度检测
为探究Fe3O4@SiO2@AgNPs的拉曼增强效果,以Fe3O4@SiO2@AgNPs为基底检测不同浓度R6G的SERS光谱。如图4所示,随着R6G浓度增大,拉曼信号显著增强,增强因子为4.27×108。

图4 探针分子R6G的SERS光谱图Fig.4 SERS spectra of Raman probe molecule R6G
2.2.2 EDTA-2Na检测的定性分析
以Fe3O4@SiO2@AgNPs为SERS基底对1 mg/mL EDTA-2Na溶液进行SERS检测,结果如图5所示。EDTA-2Na的拉曼特征峰为930、1 098、1 325、1 400 cm-1和1 625 cm-1,与Guzonas等[31]的研究结果基本一致。930 cm-1处的特征峰分别归属为C—C伸缩振动,1 098 cm-1处的特征峰归属为C—N伸缩振动,1 325 cm-1处的特征峰归属为N—H的弯曲振动,1 400 cm-1处的特征峰归属为COO—的对称伸缩振动,1 625 cm-1处的特征峰归属为COO—的不对称伸缩振动。
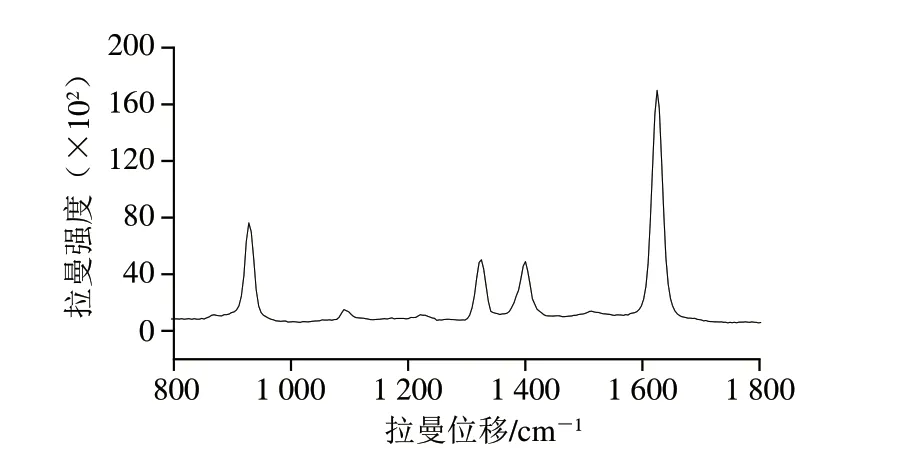
图5 1 mg/mL EDTA-2Na溶液SERS光谱图Fig.5 SERS spectra of 1 mg/mL EDTA-2Na solution
2.3 方法学验证
从图6a可知,1 625 cm-1处的特征峰强度随着EDTA-2Na标准溶液质量浓度的降低而降低,EDTA-2Na的检出限为1.75×10-6mg/mL(根据IUPAC推荐的方法即检出限=3σ/k,σ为标准偏差,k为校准曲线的斜率)。且当质量浓度在1×10-5~1 mg/mL线性范围内时,1 625 cm-1处的拉曼吸收强度与EDTA-2Na标准溶液的质量浓度对数具有良好的线性关系(图6b),线性方程为y=3 012x+16 675,R2为0.994,说明该基底可以实现对低浓度EDTA-2Na的快速分析检测。
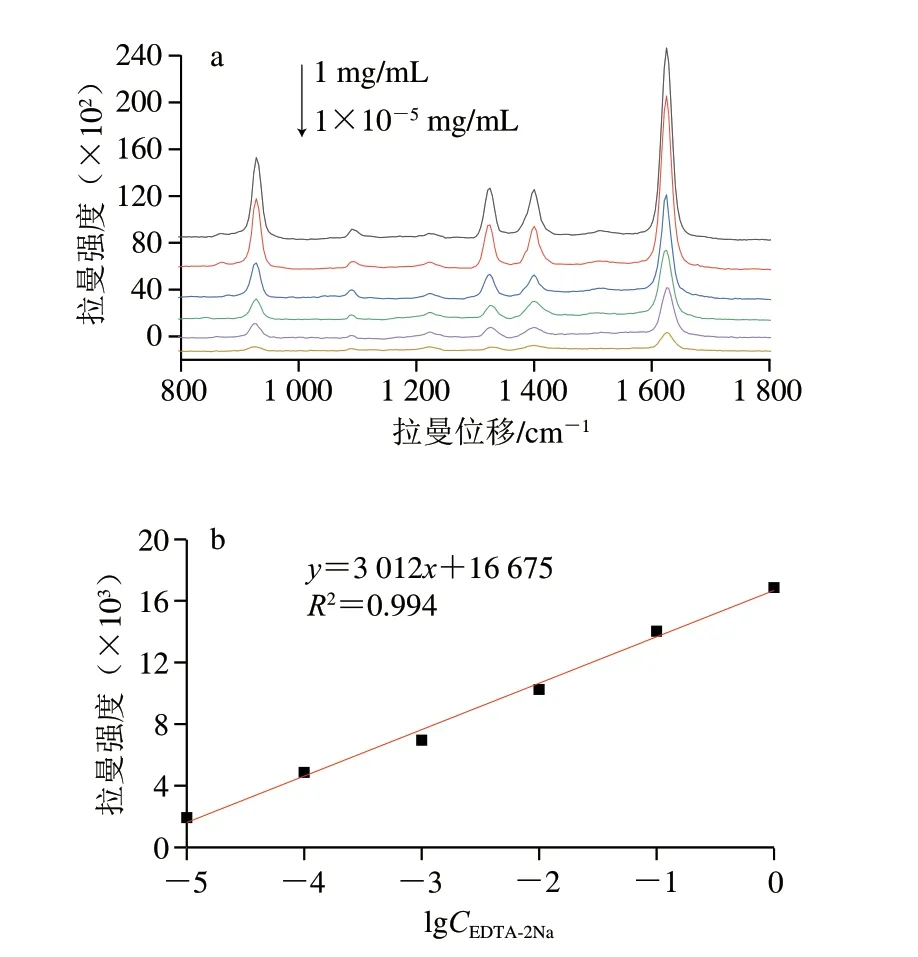
图6 不同质量浓度EDTA-2Na的SERS光谱图(a)和EDTA-2Na在1 625 cm-1处的拉曼强度与质量浓度关系图(b)Fig.6 SERS spectra of EDTA-2Na at different concentrations (a) and linear relationship between Raman intensity at 1 625 cm-1 and logarithmatic concentration of EDTA-2Na (b)
以10-5mg/mL EDTA-2Na为SERS探针,考察SERS增强基底的均一性与重复性。如图7a所示,对同一批次制备的Fe3O4@SiO2@AgNPs随机抽取6 个点进行SERS检测,以EDTA-2Na在1 625 cm-1处的拉曼信号强度计算相对标准偏差(relative standard deviation,RSD)为2.37%,显示出Fe3O4@SiO2@AgNPs良好的均一性。抽取6 组不同批次的Fe3O4@SiO2@AgNPs进行SERS检测,根据6 组材料在EDTA-2Na 1 625 cm-1处特征峰的平均拉曼信号强度,计算出RSD为4.22%。图7b的误差棒表示每组Fe3O4@SiO2@AgNPs在6 个不同测试点处拉曼强度的标准偏差,结合图7a可知,同批次的Fe3O4@SiO2@AgNPs在不同测试点的拉曼强度接近且每批次之间的拉曼强度差距不大,体现出Fe3O4@SiO2@AgNPs基底良好的均一性和重复性。
2.4 干扰实验
为考察干米粉中潜在干扰物对SERS基底检测EDTA-2Na溶液产生的影响,进行抗干扰实验。以Fe3O4@SiO2@AgNPs为SERS基底,对100 μL 1×10-5mg/mL EDTA-2Na、65 mg/mL淀粉、1.6 mg/mL纤维素、1.82×10-2mg/mL NaCl、3×10-3mg/mL KCl、0.8×10-5mg/mL抗坏血酸、2.5×10-5mg/mL Cu2+、0.5×10-5mg/mL Pb2+进行SERS检测,每个样品重复3 次取平均值,其在1 625 cm-1处的拉曼强度如图8所示。结果表明,干米粉中的潜在干扰物在EDTA-2Na 1 625 cm-1特征峰处没有拉曼峰,不会对EDTA-2Na的SERS检测产生影响,该检测方法具有良好的抗干扰能力。
2.5 预包装干米粉中EDTA-2Na的检测
抽取3 家公司生产的预包装干米粉进行前处理,采用SERS对预包装干米粉样品溶液进行检测,在样品溶液中并均未检测出EDTA-2Na的拉曼信号。采用HPLC进行辅助检测,亦未在样品溶液中检测出EDTA-2Na,SERS与HPLC检测的结果一致(表1)。证明3 种预包装干米粉样品中均未添加EDTA-2Na。

表1 预包装干米粉中EDTA-2Na的测定Table 1 Contents of EDTA-2Na in prepackaged dried rice noodle samples determined by SERS and HPLC
2.6 样品的加标回收检测
在预包装干米粉样品溶液中添加0.20~1.00 mg/kg范围内的低、中、高三个浓度的EDTA-2Na进行样品加标回收实验(图9),计算得到样品检出限为0.1 mg/kg,加标回收率在96.50%~104.67%之间,RSD在1.2%~4.2%范围内(表2)。结果表明Fe3O4@SiO2@AgNPs基底可应用于预包装干米粉中EDTA-2Na的检测,且测定结果可靠。
3 结 论
本实验建立基于SERS技术快速、灵敏检测预包装干米粉中痕量EDTA-2Na的方法。首先利用种子介导法制备了灵敏度高、重复性和均一性较好的Fe3O4@SiO2@AgNPs作为SERS基底,其对R6G的SERS增强因子为4.27×108。使用该SERS基底对预包装干米粉样品中的EDTA-2Na进行SERS检测,检出限可达0.1 mg/kg,抗干扰能力良好,加标回收率为96.50%~104.67%,RSD为1.2%~4.2%。此外,利用SERS与HPLC同时对预包装干米粉样品溶液进行检测获得了一致的结果,验证了SERS法检测预包装干米粉样品中EDTA-2Na的可靠性,证明该方法可实现对预包装干米粉样品中低浓度EDTA-2Na的快速分析检测。结果表明,利用此方法检测预包装干米粉中的EDTA-2Na切实可行,可灵敏地检测市售预包装螺蛳粉干米粉的EDTA-2Na添加情况,更好地维护预包装干米粉市场,为检测预包装干米粉中的食品添加剂——EDTA-2Na提出新的检验思路。
