双官能团化介孔二氧化硅材料的合成及其在氨基糖苷类药物检测中的应用
2023-10-21唐敏敏邵雪梅宋锦柱朱晓军许丹科
唐敏敏,邵雪梅,高 巍,宋锦柱,朱晓军,冯 云,邹 洁,*,许丹科*
(1.江苏省产品质量监督检验研究院,江苏 南京 210000;2.南京大学化学化工学院,生命分析化学国家重点实验室,江苏 南京 210046;3.南京医科大学公共卫生学院,江苏 南京 211166)
氨基糖苷类药物(aminoglycosides,AGs)是一类广谱抗生素,自1943年首次发现以来,在治疗重大细菌感染和结核病方面发挥着重要作用[1]。但是毒理学研究表明,AGs表现出相当严重的副作用,例如耳毒性和肾毒性,过敏反应。牛奶是日常饮食中的常见食物,含有脂肪、多肽、蛋白质和矿物质元素等多种营养物质。AGs具有广谱抗菌性,可治疗奶牛乳腺炎、奶牛结核病等,还可作为促生长剂提高饲料报酬率,因此被直接或间接使用于奶和奶制品的生产、加工和贮藏中[2]。人类食用这些有药物残留的动物产品也会遭受风险,此外,还易导致细菌耐药性的发展,从而给疾病治疗增加难度。GB 31650—2019《食品中兽药最大残留限量》中规定了AGs在不同动物组织中的最大残留限量,规定在牛奶中庆大霉素不得超过200 μg/kg,卡那霉素不得超过150 μg/kg,新霉素不得超过1 500 μg/kg。为规范AGs使用,政府每年均需要投入大量的人力物力保证农产品安全。因此,建立具有高准确度和灵敏度的AGs的检测方法尤为重要。
固相萃取(solid phase extract,SPE)在兽药前处理中发挥相当重要的作用,不仅能够富集目标物,还可以净化样品基质。牛奶中AGs中最常用的是C18反相SPE[3]或MCX、WCX离子交换模式的SPE[4-7]。与单模式吸附剂相比,混合模式吸附剂通过多种相互作用具有更好的吸附选择性,可以通过控制洗涤条件轻松去除杂质。除了常规使用的SPE柱外,不少研究者利用分子印迹[8-9]或磁珠、石墨烯等材料键合硼酸基团[10]、羟基[11-12]、羧基[13]开发了一系列AGs的吸附剂。介孔材料MCM-41型材料的骨架由六边形排列的圆柱形介孔组成,具有高表面积和窄孔径分布,优异的孔隙结构,高的热稳定性和化学稳定性,其表面具有大量的硅羟基,可通过官能团的修饰增加功能,在吸附化学领域引起了极大的研究兴趣[14]。目前已有文献报道表面修饰的介孔材料在铅、阴离子有毒偶氮染料、生物毒素等方面的应用[15-18],但在AGs富集上还应用较少。
由于AGs不具有挥发性,没有发色团或者荧光团,不适合直接进行紫外或荧光检测[1],需要进行衍生处理,这增加了前处理的复杂性。随着质谱技术的出现,液相色谱-串联质谱技术成为最简单和准确的检测手段[2]。在色谱分离上,AGs是具有高极性的化合物,它们在普通反相色谱柱上的保留相当弱[13]。最常见的解决方法是在流动相中添加离子对试剂[10]。但是,这些添加剂可能会导致色谱柱严重污染和电喷雾电离(electrospray ionization,ESI)的离子抑制作用[19]。Amide柱为酰胺基键合亚乙基桥杂化颗粒色谱柱,较C18反相柱、亲水型的HSS T3和BEH HILIC正相柱,具有易保留与分离强极性分析物的特点,同时可避免离子对试剂对仪器的污染,因此比离子对色谱法更有优势。
本研究基于介孔材料MCM-41开发了一种新的反相/阳离子交换混合模式的吸附剂,以安普霉素(apramycin,APC)、庆大霉素(gentamicin,GTC)、卡那霉素(kanamycin,KNM)、新霉素(neomycin,NMC)、阿米卡星(amikacin,AKH)和妥布霉素(tobramycin,TBC)6 种AGs为目标分析物。合成的双官能团化的吸附剂中含有丰富的羧基,在合适的pH值下可解离,预期通过阳离子交换作用有效地提取AGs,同时,合成材料和AGs有一定的疏水相互作用,可通过调整淋洗液洗去杂质,进一步实现净化。开发的吸附剂对目标物有较好的吸附能力和良好的选择性,并进一步评估其在超高效液相色谱-串联质谱(ultra-high performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)分析中对基质效应(matrix effect,ME)的改善效果。
1 材料与方法
1.1 材料与试剂
全脂牛奶样品 江苏南京苏果超市。实验前经过国家标准方法(GB/T 22969—2008《奶粉和牛奶中链霉素、双氢链霉素和卡那霉素残留量的测定 液相色谱-串联质谱法》)测定不含6 种AGs中的任何一种,作为基质空白样品用于ME和方法开发研究。
MCM-41 江苏先丰纳米材料科技有限公司;十八烷基三甲氧基硅烷(octadecyltrimethoxysilane,C18TMS) 上海泰坦科技股份有限公司;无水甲苯、N,N-二甲基甲酰胺(N,N-dimethylformamide,DMF)南京化学试剂股份有限公司;3-氨丙基三乙氧基硅烷(3-aminopropyltriethoxysilane,APTES)、琥珀酸苷(succinic acid glycoside,SAA)、三氯乙酸、磷酸氢二钾 阿拉丁化学试剂有限公司;无水乙醇、氢氧化钠国药集团化学试剂有限公司;以上试剂均为分析纯。AGs标准品 上海安谱实验科技股份有限公司(生产商为德国Dr.EhrensorferGmbH公司)。
1.2 仪器与设备
Arium Pro超纯水系统 德国Sartorius公司;ME104E万分之一电子分析天平 瑞士Mettler Toledo公司;5810R高速冷冻离心机 美国Eppendorf公司;DZF-6051真空干燥箱 南京市南北仪器公司;Nicolet Is50傅里叶变换红外光谱仪 美国Thermo Scientific公司;JEOL JSM-7800F热场扫描电子显微镜、JEOL 2800高通量投射电子显微镜 日本电子公司;JWBK200B比表面积和孔径分析仪 北京精微高博科技有限公司;API 6500 Plus三重四极杆串联质谱仪(搭载Sciex Exion LC超高效液相色谱) 美国AB SCIEX公司。
1.3 方法
1.3.1 C18/COOH功能化介孔MCM-41材料的合成
1.3.1.1 C18/NH2功能化介孔MCM-41材料的制备
根据文献[20]的方法进行合成,并作修改。具体如下,将1 g MCM-41分散于100 mL无水甲苯中,在60 ℃机械搅拌20 min,随后逐滴加入APTES和C18TMS各1.2 mL,于110 ℃油浴中剧烈搅拌回流15 h,将所得的白色固体产物收集,并用无水乙醇洗涤3 次,于烘箱中真空干燥(真空度-0.08 MPa,65 ℃,12 h)得到C18/NH2功能化的介孔MCM-41材料(MCM-41@NH2@C18)。同时,对照材料分别为不加入APTES和C18TMS,仅加入APTES,其余处理方法相同。
1.3.1.2 C18/COOH功能化介孔MCM-41材料的制备
羧基功能化的方法参照文献[21],并作修改。称取C18/NH2功能化介孔MCM-41材料0.8 g,分散于100 mL DMF中,室温搅拌20 min。称取11.5 g SAA溶于50 mL DMF中,逐滴缓慢滴入搅拌均匀的反应体系中,滴加完之后继续室温搅拌24 h。反应结束后,收集白色粉末,并用无水乙醇洗涤3 次,于烘箱中真空干燥(65 ℃、12 h)得到最终反应产物(MCM-41@COOH@C18)。制备流程示意图见图1。

图1 MCM-41@COOH@C18的合成示意图和检测流程图Fig.1 Schematic diagram for the synthesis of MCM-41@COOH@C18 and its application in SPE
1.3.2 样品制备
准确吸取牛奶样品1.0 mL,加入5%三氯乙酸溶液10 mL和0.1 mol/L磷酸氢二钾溶液5 mL,于50 mL塑料离心管中混合。涡旋30 s,振荡5 min。在4 ℃、10 000 r/min离心2 min后,将上清液转移到另一50 mL离心管中,用10 mol/L NaOH溶液调节pH值至6.0,备用。
1.3.3 SPE步骤
将MCM-41@COOH@C18介孔微球(30 mg)装填至两端具有筛板的SPE小柱中(3 mL)。使用前预先用3 mL甲醇和3 mL水活化填料。将上述提取溶液全部通过SPE小柱后,依次用2 mL水和2 mL甲醇淋洗,抽干。用1 mL水-乙腈-甲酸(93∶5∶2,V/V)洗脱,收集洗脱液,在UPLC-MS/MS分析之前使用0.22 μm滤膜过滤,SPE过程示意图见图1。
1.3.4 回收率实验
日内回收率实验:在空白牛奶样品中分别加入5、50、200 ng/mL的AGs标准品,每个质量浓度做6 次重复。按照建立的方法进行实验,计算回收率和精密度。
日间回收率:在空白牛奶样品中分别加入5、50、200 ng/mL的AGs标准品,每个浓度做6 次重复,按照建立的方法测试3 d,计算回收率和精密度。
1.3.5 UPLC-MS/MS检测
在Waters ACQUITY BEH Amide(2.1 mm×100 mm,1.7 μm)上进行色谱分离,流动相A为2 mmol/L乙酸铵溶液(1%甲酸),流动相B为2 mmol/L乙酸铵乙腈溶液(1%甲酸)。线性梯度为:0~2.5 min,20% A、80% B;2.5~3.5 min,20%~65% A、80%~35% B;3.5 ~5.5 m i n,6 5%~9 0% A、3 5%~1 0% B;5.5~6.5 min,90% A、10% B;6.5~6.6 min,90%~20% A、10%~80% B;6.6~9 min,20% A、80% B。流动相流速为0.3 mL/min。柱温设置为40 ℃,进样量为2 μL。
通过使用注射泵以10.0 μL/min的流速连续注入AGs混合标准溶液(100.0 ng/mL),优化ESI+的质谱参数。MS条件:ESI+模式,离子源电压5.5 kV;雾化温度550 ℃;雾化气(空气)压力50 psi;辅助气(空气)压力55 psi;气帘气(氮气)压力45.0 psi。在多反应监测(multiple reaction monitoring,MRM)扫描模式下进行采集。选择质子化的分子离子作为母离子,使用两个子离子进行定量和定性。MRM模式质谱参数如表1所示。
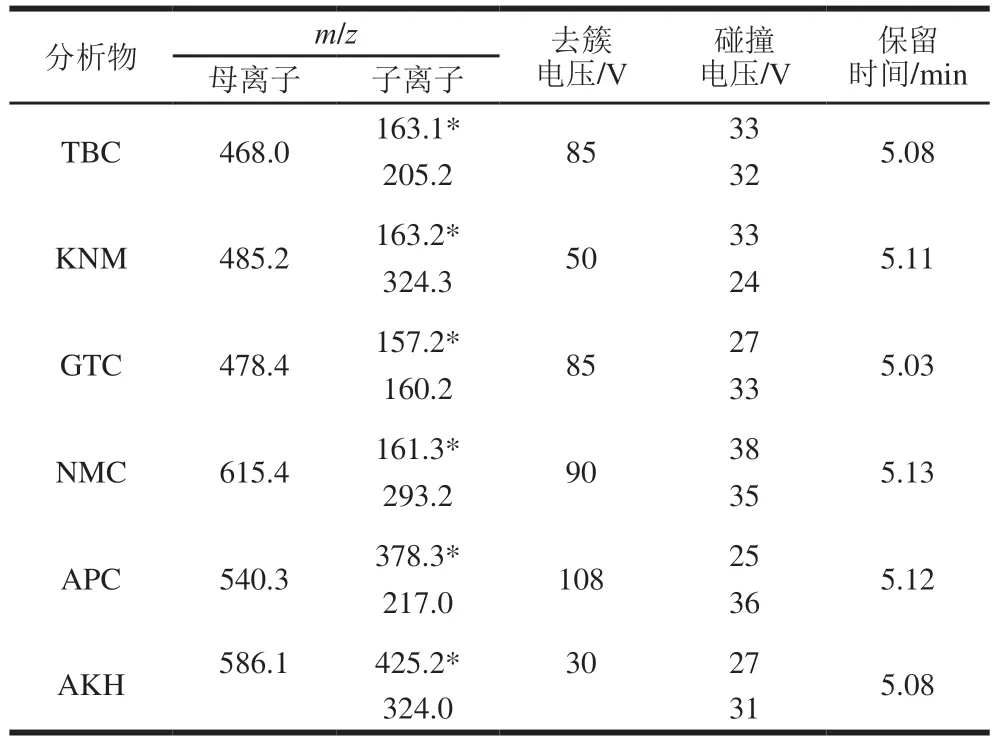
表1 MRM模式下AGs的质谱优化参数Table 1 Optimized MS parameters for AGs in MRM mode
1.3.6 标准溶液配制
标准储备液配制:准确称取适量标准品于10 mL聚丙烯容量瓶中,用水溶解后定容,配制成0.5 mg/mL的标准储备溶液。
混合标准中间溶液配制:分别吸取5 mL标准储备溶液置于50 mL聚丙烯容量瓶中,用水-乙腈-甲酸(93∶5∶2,V/V)定容,配制成50 μg/mL的中间液。
混合标准工作溶液配制:溶液标准曲线用水-乙腈-甲酸(93∶5∶2,V/V)将混合标准中间液稀释至5.0、10.0、20.0、50.0、100.0、500.0 ng/mL的一系列质量浓度。基质匹配标准溶液用基质空白溶液稀释至相同质量浓度。
1.4 数据统计分析
使用SPSS 19.0统计软件进行数据分析处理,采用Microsoft Excel 2010进行图表绘制。
2 结果与分析
2.1 吸附材料的表征
由于MCM-41介孔材料具有均一孔径的长程有序的孔道结构和极高的比表面积,非常适合作为吸附材料[14]。所制备的MCM-41@COOH@C18不破坏原有的孔道结构,在SEM(图2a)中可以清楚地观察到蠕虫状的颗粒,呈簇状生长,与Zhang Jiaojing等[22]的报道一致,排列致密,最小粒径约为200 nm。此外,如图2b所示,TEM显微图片证明了高度有序的介孔结构,显示了约4 nm宽的有序通道的存在。

图2 MCM-41@COOH@C18的扫描电镜图(a)和透射电镜图(b)Fig.2 SEM (a) and TEM (b) micrographs of MCM-41@COOH@C18
通过氮气吸附脱附技术,MCM-41和MCM-41@COOH@C18两种材料的吸附-脱附曲线和孔径分布如图3所示。根据IUPAC分类,这两条曲线为IV型。这种等温线是介孔材料的典型特征曲线[23]。MCM-41的BET比表面积为1 049.9 m2/g,平均孔径为3.6 nm。从图3a可以看出,相对压力(P/P0)<0.3时,氮的吸附量随着P/P0增加而增加。这是因为在初始阶段发生了单层吸附。P/P0为0.3~0.4时,由于氮气在介孔材料中发生毛细管团聚,氮气吸附量急剧上升。当P/P0为0.4~1时,氮吸附量增长缓慢,说明材料外比表面积较低,孔隙数量较少。引入—C18和—COOH基团时,样品表现出不那么尖锐的台阶,并且MCM-41@COOH@C18的孔径显示出了双峰孔分布,双官能团修饰后材料的比表面积和孔径都有一定程度的减小,比表面积为505.8 m2/g,平均孔径为2.6 nm,这归因于修饰的基团占据了结构通道。

图3 N2吸附-脱附等温线和孔径分布曲线Fig.3 N2 adsorption-desorption isotherm and pore size distribution curves of MCM-41 and MCM-41@COOH@C18
为了验证合成的吸附剂有效基团,图4显示了MCM-41原材料和合成后的MCM-41@COOH@C18的傅里叶变换红外光谱。MCM-41和MCM-41@COOH@C18在808、978 cm-1和1 088 cm-1处显示出二氧化硅的典型特征峰,分别对应Si—O—Si对称键、Si—OH伸缩键、Si—O—Si不对称键的振动。光谱中观察到的3 445、3 550 cm-1和1 632 cm-1,1 651 cm-1处的吸附带为硅烷醇基团的—OH拉伸[22]。由于C18和羧基基团的修饰,合成后的MCM-41@COOH@C18在2 855 cm-1和2 928 cm-1处出现两个新峰,这是由于C18链上—CH2—和—CH3的C—H振动[18]。1 724 cm-1的新峰是羧基的C=O峰,这些结果证实了MCM-41表面改性的成功。

图4 MCM-41, MCM-41@COOH@C18的傅里叶红外光谱图Fig.4 Fourier infrared spectra of MCM-41 and MCM-41@COOH@C18
2.2 SPE条件的优化
以上结果证明成功合成了该介孔材料,制备好的MCM-41@COOH@C18材料具有良好的水分散性,可以在水溶液中均匀分散,没有任何聚集。选择的6 种AGs化学结构中均有许多氨基和羟基,属于亲水性的化合物。预测富集机理是反相和离子交换两种模式共同决定的,由于MCM-41@COOH@C18含有丰富的羧基,羧基在合适的pH值条件下可解离,可通过阳离子交换有效地富集AGs,同时,键合的C18和AGs有一定的疏水相互作用,可以利用淋洗液的选择进一步分离目标物和杂质。为评估MCM-41@COOH@C18介孔材料在富集AGs的适用性,进一步优化可能影响提取的参数,例如提取液种类、提取时的pH值(3.0~7.0)、吸附剂的量、洗脱溶剂种类、洗脱溶剂体积。
2.2.1 提取液种类选择
常用的AGs提取溶剂有磷酸盐缓冲溶液、三氯乙酸、乙二胺四乙酸二钠盐溶液。对这3 种溶液的提取效果分别进行比较,发现用磷酸盐缓冲液和乙二胺四乙酸二钠处理后的样品提取液浑浊,影响后续SPE柱净化和回收率实验。三氯乙酸提取后,离心后的溶液较为澄清,没有絮状杂质。说明三氯乙酸易于沉淀牛奶中的蛋白质,同时加入的磷酸盐缓冲液有助于pH值的调节,因此最终选择5%三氯乙酸溶液10 mL和0.1 mol/L磷酸氢二钾溶液5 mL作为提取溶液。
2.2.2 pH值的优化
所制备的MCM-41@COOH@C18具有大的比表面积、均匀的孔径和良好的双官能团修饰。经过SPE时,备用溶液的pH值会影响目标物的离子化。合成的双功能化吸附剂中含有丰富的羧基,在适宜的pH值下可解离,与AGs上的氨基发生离子交换作用,因此比较了备用溶液的pH值为3、4、5、5.5、6、6.5、7时,目标物在SPE柱上的保留情况。富集前牛奶中的AGs质量浓度为100 ng/mL,保证同样的SPE柱填充量为30 mg,将通过SPE柱的不同p H 值的样品溶液收集,用G B/T 2 2 9 6 9—2 0 0 8 进行检测(预先经过实验验证,该方法回收率范围86.9%~98.5%),得到富集前后备用溶液中的AGs含量。图5表明,当溶液pH值为3和4时,每种目标药物在合成的吸附填料上的富集率均不超过50%,当pH值为5和5.5时,AKH、KNM的富集率不超过80%,当溶液pH值为6~7时,溶液中的AGs在SPE柱上的富集率较高。这说明样品pH值是影响离子交换模式的关键因素,综合考虑,选定样品溶液的pH值为6,此时目标物的富集率为84.0%~97.6%。
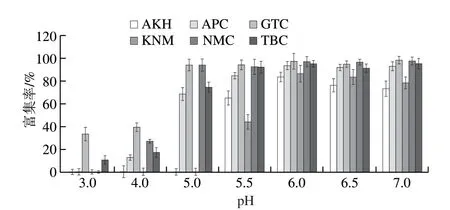
图5 样品pH值对富集效率的影响Fig.5 Effect of pH on enrichment efficiency
2.2.3 SPE柱填料用量的选择
因为吸附剂承载目标物的能力有限,为了获得最大的富集效果,足够的吸附剂用量必不可少,选择SPE柱填充量为30 mg,比较不同质量浓度(40、80、400 ng/mL和2 000 ng/mL)的氨基糖苷溶液的回收率情况(图6a),发现对于每一种AGs,不同质量浓度的相应目标物均能取得较为一致和稳定的回收率,且回收率范围均在70%~110%之间,满足分析方法要求,说明30 mg的填料含量不会存在柱过载现象,因此选择SPE填料量为30 mg。
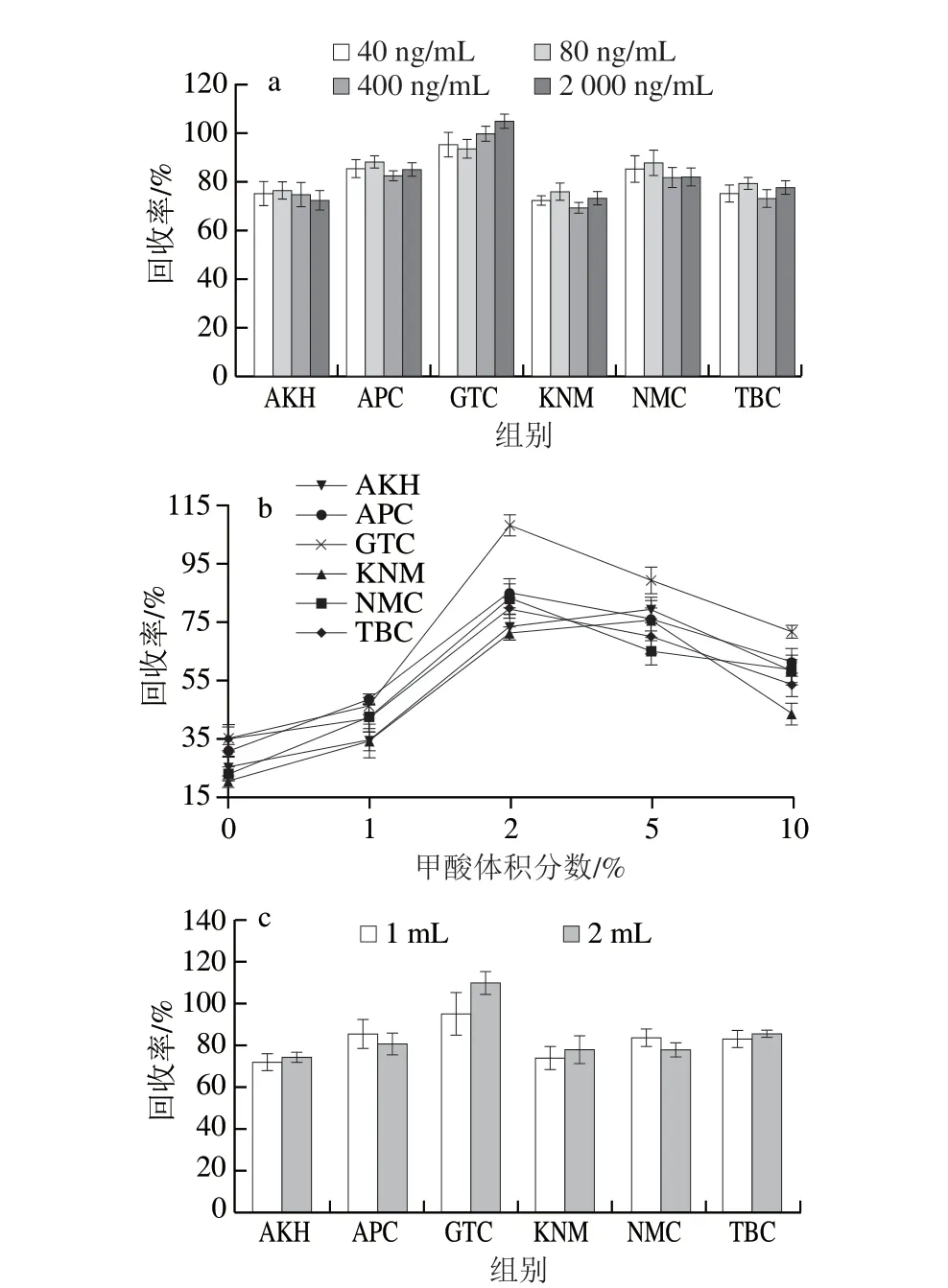
图6 不同条件对回收率的影响(n=3)Fig.6 Effect of SPE conditions on recovery (n = 3)
2.2.4 淋洗液的选择
淋洗液在一个完整的SPE过程中可洗涤富集在SPE填料上的共萃取物,有效将目标物和干扰组分分离。理想的淋洗液应不损失目标物同时又可洗涤杂质,减少基质干扰。选择2 mL水和2 mL甲醇两种不同性质的溶剂作为淋洗液,收集后经过UPLC-MS/MS检测,均没有目标物存在,说明淋洗液不会造成目标物的损失。
2.2.5 洗脱溶剂的选择
AGs分子中具有丰富的氨基,可以与合成的MCM-41@COOH@C18中的羧基产生阳离子交换作用,在洗脱溶剂中加入酸会有助于削弱阳离子交换相互作用[24]。AGs又是极性较高的一类药物,因此在洗脱溶剂的选择上,选择乙腈、水和甲酸的混合物为洗脱溶剂,其中,乙腈的比例不宜过高,过高的乙腈含量会导致目标药物和吸附剂之间的亲水相互作用不易被破坏。设定为在整个洗脱体系中乙腈体积分数为5%,酸溶液的选择既能提供一定的酸度,也要适用于质谱离子源的离子化,因此选择挥发性酸甲酸。选择提取液的pH值为6,SPE填料量为30 mg,水和甲醇为淋洗液,比较甲酸体积分数从0%到5%以选择最优的洗脱溶剂。从图6b可以看出,当甲酸体积分数为2%时,6 种AGs回收率均超过70%。因此,选择洗脱液比例为水-乙腈-甲酸(93∶5∶2,V/V)。
2.2.6 洗脱液体积优化
除了洗脱溶剂的类型,洗脱溶剂的体积也影响着整个SPE的回收率。比较水-乙腈-甲酸(93∶5∶2,V/V)1 mL和2 mL对目标物回收率的影响,其他参数保持相同。如图6c所示,这两种洗脱体积对目标物的回收率并没有造成显著性差异,1 mL的洗脱液用量回收率均超过70%,说明1 mL的洗脱用量足够洗脱AGs。洗脱体积增加,造成目标物的稀释,还需要采取后续的浓缩步骤,会增加实验的复杂程度。因此选择洗脱液用量为1 mL。
2.3 ME评估
在HPLC-MS/MS经常观察到信号增强或抑制效应,导致回收率偏高或者偏低[25]。牛奶中含有一些内源性物质,如蛋白、脂质、氨基酸和其他复杂成分[10],ME可通过基质匹配标准曲线和溶液标准曲线评估。ME/%=空白基质中标准物质的峰面积/溶剂中标准物质的峰面积×100。当ME在80%~120%范围内时,ME被认为可以忽略。ME大于120%表示信号增强,而ME小于80%表示信号抑制[6,26-27]。不使用任何净化方式,直接将备用液进行UPLC-MS/MS检测,会导致ME值很低(42.0%~62.0%)(图7),表明存在信号抑制。相比之下,使用仅经过羧基修饰的MCM-41@COOH材料,部分药物的ME值得到了改善,如AKH、KNM的ME值在80%~120%的合理范围内,但GTC的ME值高达229.9%。在羧基化填料的基础上,同时引入了C18基团,这时目标物产生的ME值范围为78.0%~120.0%,说明双官能化的MCM-41@COOH@C18的材料在基质净化方面更有优势。将合成的MCM-41@COOH@C18吸附剂与商业产品WCX(3 mL/60 mg,Waters)进行比较。如图7所示,由于WCX SPE柱是阳离子交换柱,它的ME值为72.0%~114%,主要是因为它可以选择性地吸附碱性目标物,而用水和甲醇洗涤可以排除中性和脂类化合物。因此,混合模式SPE在降低ME方面与商业WCX SPESPE柱相当,说明合成的MCM-41@COOH@C18具有显著的改善ME的效果。从理论上讲,混合模式吸附剂降低ME的能力与大量疏水性、中性等杂质有关,这些杂质可以通过用水和甲醇洗涤去除。

图7 不同SPE填料对ME的影响Fig.7 Effect of different SPE fillers on matrix effect
2.4 色谱、质谱条件优化
色谱分离上,由于AGs是强极性化合物,在普通反相色谱柱上的保留相当弱。为了在反相色谱柱上有较好的保留,常会在流动相中加入三氟乙酸等离子对试剂[28],但是离子对试剂会造成基质抑制,损害柱寿命等问题,因此AGs的分离也是检测人员面临的挑战之一。近年来,氨基柱也被广泛用于AGs的分析[29]。该柱在亚乙基桥杂化的高纯硅胶颗粒上键合了酰胺基,相较于HILIC柱具有更高的稳定性。本实验用的是Waters ACQUITY BEH Amide(2.1 mm×100 mm,1.7 μm)色谱柱,其适用于分离强极性物质。考察纯水、甲酸溶液、乙酸溶液、乙酸铵缓冲液作为水相,乙腈作为有机相对AGs的分离效果,发现2 mmol/L的乙酸铵溶液的峰形和灵敏度最佳。在流动相中加入1%的甲酸有助于增强离子化程度,提高灵敏度。在有机相中加入2 mmol/L乙酸铵时,稳定的酸性环境增强了对目标物的洗脱能力,减少拖尾现象[30]。从图8可以看出,牛奶基质中添加100 ng/mL AGs的色谱图,目标峰附近没有明显的干扰,峰形和响应良好,这表明该实验条件下可以分离6 种AGs。
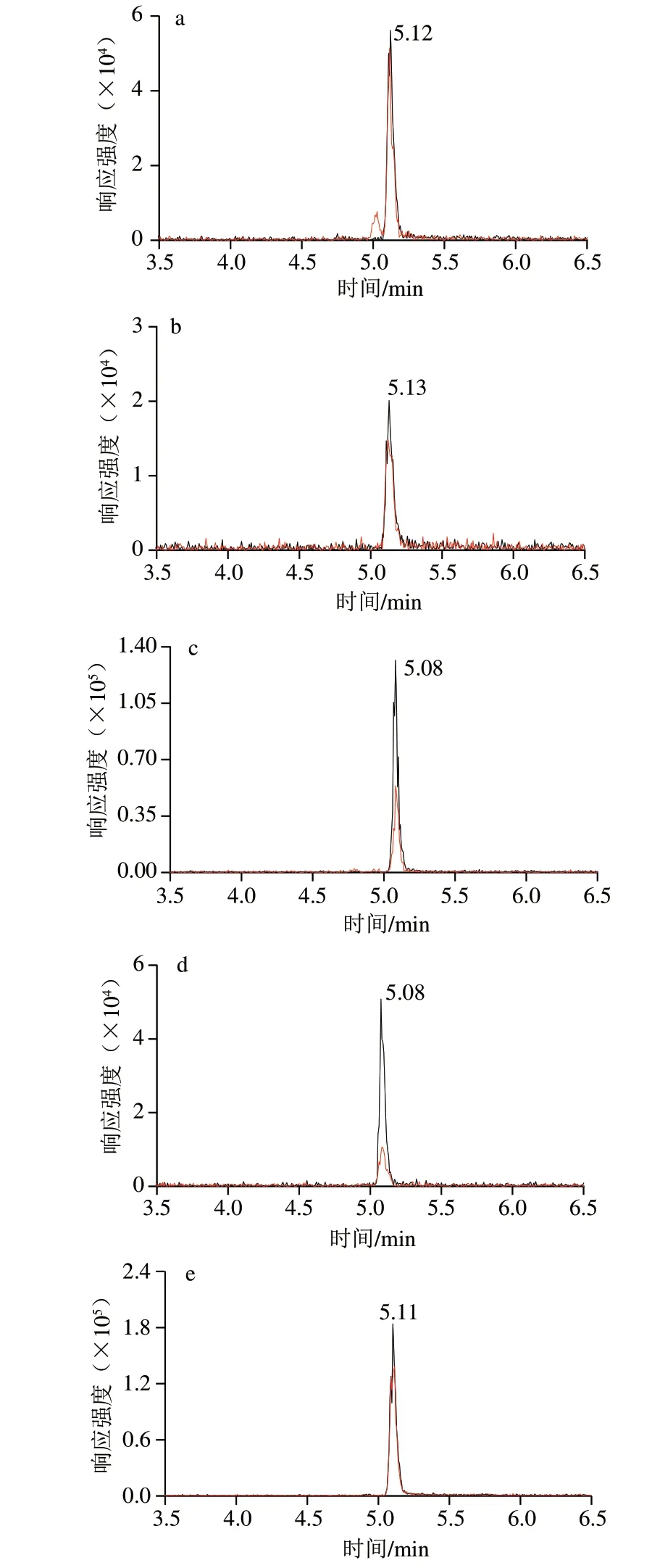
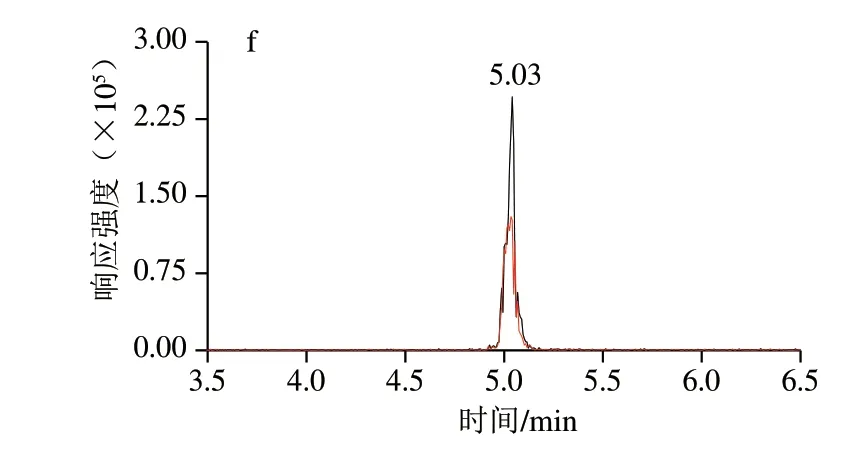
图8 6 种AGs的提取离子流图Fig.8 Extracted ion current chromatograms of six AGs
AGs结构中含有氨基环醇、羟基和伯胺或仲胺基团等,呈强极性和弱碱性,适合ESI+扫描模式。选择100 ng/mL混合标准溶液连续注射进入质谱离子源入口,优化后的质谱条件见表1。
2.5 方法验证
采用基质匹配标准曲线进行定量,标准曲线质量浓度为5.0、10.0、20.0、50.0、100.0 ng/mL和500.0 ng/mL,线性方程见表2,相关系数(r)为0.992 7~0.999 5,均呈现良好的线性关系。

表2 6 种AGs的线性方程、相关系数、检出限和定量限Table 2 Linear equations, correlation coefficients, LODs and LOQs of six AGs
基于信噪比为3,牛奶中AGs检出限为0.30~2.50 ng/mL,基于信噪比为1 0 , 牛奶中A G s 定量限为1.00~10.0 ng/ mL,这完全符合牛奶中AGs检测的要求。使用低、中、高质量浓度(5、50、200 ng/mL)的加标空白样品进行回收率实验考察,结果见表3。每个水平做6 次重复,6 种AGs的平均日内回收率范围为74.3%~107%,相对标准偏差(relative standard deviation,RSD)范围分别为3.0%~13.3%;日间回收率范围为77.2%~106%,RSD范围为2.6%~15.7%。这些结果表明,将合成的MCM-41@COOH@C18填料应用于牛奶中的AGs检测的准确度和精密度能够满足常规监测的要求。
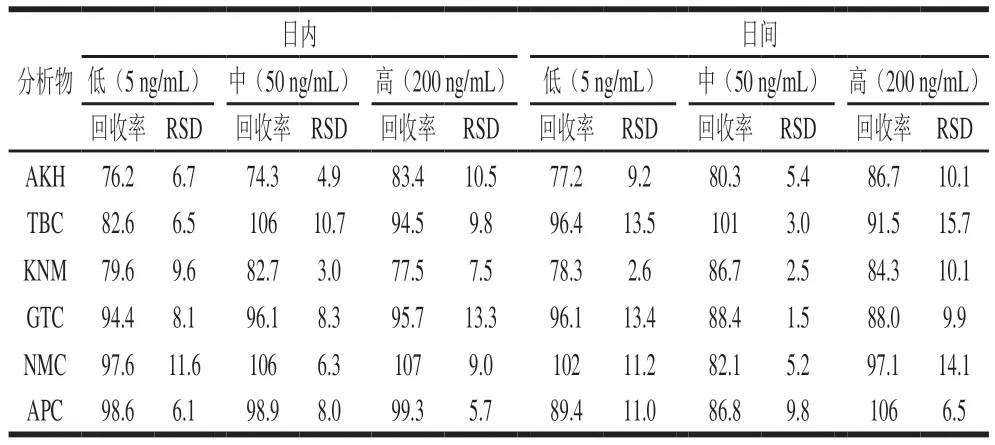
表3 牛奶中添加3 个水平的AGs的回收率和重复性(n=6)Table 3 Recoveries and repeatability RSDs of AGs at different concentrations in spiked milk (n = 6)%
2.6 与已经报道的方法的比较
将所建立的方法与近年来报道的不同基质样品中的AGs的其他技术进行比较(表4)。报道的文献主要基于SPE、MDSPE与MS/MS检测系统的耦合。无论使用哪种SPE类型,报道的吸附剂都是基于分子印迹材料,反相或离子交换的,仅针对单独吸附模式。相比之下,反相/阳离子交换型吸附剂与AGs的结构相匹配,因此可以通过静电基团选择性地吸附目标物,同时利用反相作用有效分离目标物和杂质。在材料制备上,原料成本低廉,合成方式简单;在目标物检测方面,所建立的方法覆盖种类多,能够同时有效测定6 种AGs,说明合成的MCM-41@COOH@C18对AGs具有良好的吸附和洗脱特性;在方法性能上,AKH、TBC、KNM、GTC、NMC、APC的回收率均在74.3%~107%之间,方法检出限在同类样品检测中也优于其他已报道的方法。
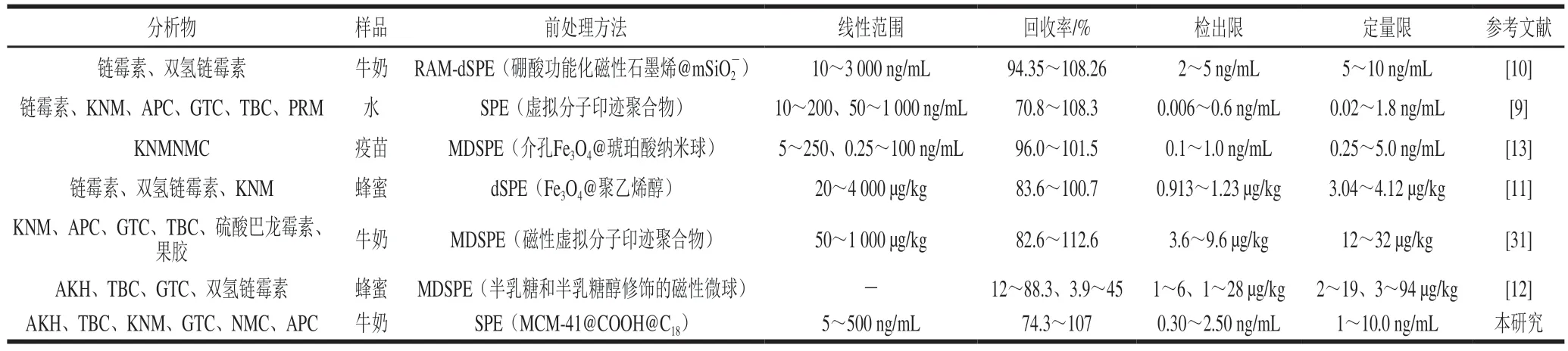
表4 与其他报道的AGs LC-MS/MS检测方法的比较Table 4 Comparison of the proposed method with other reported methods for detection of AGs
3 结 论
通过两步反应法合成了MCM-41@COOH@C18介孔材料,并将其作为SPE吸附剂填料用于牛奶中AGs的残留分析。基于介孔材料的大的外表面积和介孔通道,对其进行羧基和烷基的双官能团修饰。由于材料上的羧基和氨基之间的离子交换相互作用,通过调节淋洗液将样品基质进行了净化,显示了该合成材料在复杂基质中选择性富集和纯化AGs的巨大能力。为了获得最佳SPE富集效果,进一步系统优化SPE条件,结果表明在5~500 ng/mL线性范围内具有良好的回收率和精密度。该方法简便、经济、灵敏,可操作性强,已将其成功应用于牛奶样品中6 种AGs的提取。未来可将该MCM-41@COOH@C18介孔材料作为吸附剂拓展到其他食品基质中。
