双重荧光RT-ERA技术快速检测贝类食品中诺如病毒
2023-10-21吴占文李红娜杨艳歌
吴占文,王 帅,康 婕,李 涛,李红娜,蔡 杰,张 昊,杨艳歌,*,袁 飞,*
(1.中国检验检疫科学研究院,国家市场监管重点实验室(食品质量与安全),北京 100176;2.黑龙江八一农垦大学生命科学技术学院,黑龙江 大庆 163000;3.陕西省产品质量监督检验研究院,陕西 西安 710048)
以食物和水作为传播基质的诺如病毒(norovirus,NoV)是世界范围内导致急性胃肠炎爆发及散发的主要病原体[1]。这是由于NoV具有极强的感染性,1 个NoV颗粒引发的感染率可达50%,通常只要10 个病毒颗粒就可以使人致病,超过了已报道的任何一种食源性病毒[2-5]。据世界卫生组织(World Health Organization,WHO)报道,NoV每年可造成6.85亿腹泻病例,21.25万 人死亡[6-9],其中99%发生于发展中国家[10-12]。由于WHO已开展大规模轮状病毒疫苗接种,使NoV成为了当前儿童急性胃肠炎的主要病因[13-14]。例如秘鲁的一项研究发现,当地36%的婴儿成长至2 岁时,都会经历5 次以上的NoV感染[15],这无疑为当地食品安全检测行业敲响了警钟。近几年我国儿童急性胃肠炎病例爆发的频率不断增加[16-17],NoV的防治也变得尤为重要。但遗憾的是,目前仍未研发出有效的NoV疫苗[18]。因此,如何快速检测食物与饮用水中的NoV,并由此阻断NoV的传播,仍然是研究人员不断探索的问题。
NoV传播与食品污染密切相关,尤其是海洋中的贝类生物,海产贝类因其底栖和滤食性特性,可在海洋环境中不断对NoV进行富集[19-20]。有研究表明,以牡蛎为代表的贝类生物体内存在着与NoV结合的特定受体[21],这使NoV一旦进入其体内,很难通过自身代谢与净化作用排出体外,导致一些贝类生物体内的病毒浓度比周围环境高几十甚至几千倍[22-23]。而人们有时会直接生食贝类生物,这无疑大大增加了NoV感染的风险[24-25]。因此,加强贝类生物中NoV快速检测方法的研究,对于保障食品安全与民众健康具有重要意义[26]。
逆转录-酶促重组等温扩增技术(r e v e r s e transcription-enzymatic recombinase amplification,RTERA)是近些年新研发的一种等温核酸扩增技术[27],RT-ERA可以在37~42 ℃恒温条件下,将微量RNA的特异性区段在数分钟内扩增数十亿倍,具有优秀的应用前景。与逆转录聚合酶链式反应(reverse transcriptionpolymerase chain reaction,RT-PCR)相比,无论在检测时间还是仪器限制方面都具有显著的优势。而MS2噬菌体与NoV大小结构相似,且两者均为单链RNA病毒,可以作为过程控制模式病毒添加到贝类生物样品中[28]。基于此,本研究以MS2噬菌体作为过程控制病毒,建立NoV双重荧光RT-ERA快速检测方法,并实现了对MS2、GI和GII NoV RNA的便携式、可视化、无热循环的快速检测,检测时间仅需10 min,灵敏度可达102个拷贝,方案详见图1(使用Figdraw绘制)。

图1 NoV检测方案Fig.1 Scheme of NoV detection
1 材料与方法
1.1 材料与试剂
MS2噬菌体标准样品由中国食品药品检定研究院惠赠,GI和GII诺如病毒粪便样本由中国检验检疫科学研究院卫生检验与检疫研究所提供,轮状病毒与甲型肝炎病毒RNA样品由中国海关科学技术研究中心惠赠。贝类市售样品采集于北京、天津、辽宁大连水产市场。
RT-荧光型核酸扩增试剂盒(ERA法) 苏州先达基因科技有限公司;HiScriptII One Step qRT-PCR Probe Kit、T7 High Yield RNA Transcription Kit、FastPure Cell/Tissue Total RNA Isolation Kit V2 诺唯赞生物科技有限公司;MS2过程控制试剂盒(RT-PCR探针法-50 ℃)美正生物科技有限公司;PureLink™ RNA Mini Kit、UltraPureTM无DNase/RNase蒸馏水 美国Invitrogen公司;E.Z.N.A.®Viral RNA Kit 北京冬歌博业生物科技有限公司;Qiagen RNeasy Mini Kit 凯杰企业管理(上海)有限公司;蛋白酶K 德国默克公司;NdeI/XhoI限制性内切酶NEB(北京)有限公司;GI和GII NoV靶基因合成 北京六合华大基因科技有限公司。
1.2 仪器与设备
ABI 7500实时荧光PCR仪 美国Applied Biosystems公司;Pico17离心机 美国Thermo Scientific 公司;迷你qPCR仪 上海翊辉生物科技有限公司;智能化超微量核酸检测仪 日本岛津公司;移液器、混匀仪 德国Eppendorf公司;生物安全柜 美国NuAire公司。
1.3 方法
1.3.1 RNA标准品的构建与鉴定
分别选取GenBank数据库中GI NoV——诺瓦克病毒(Norwalk virus)(GenBank: M87661.2)和GII NoV——Lordsdale病毒(GenBank: X86557.1)位于ORF1-ORF2连接区域处的基因序列,并在序列的5’端加入酶切位点NdeI,3’端加入酶切位点XhoI,委托北京六合华大基因科技有限公司进行人工合成,并将合成片段克隆到含有T7启动子的pET28-a+载体中。挑选单克隆进行质粒提取,采用测序的方法对阳性质粒进行鉴定。将测序结果均符合预期的质粒分别命名为pET-NoV GI与pET-NoV GII。
使用酶切的方法对质粒进行线性化处理,酶切体系:阳性质粒20 μL、XhoI 1 μL、10×NEBuffer 5 μL、焦碳酸二乙酯(diethyl pyrocarbonate,DEPC)处理过的无菌ddH2O 24 μL;酶切条件:37 ℃,15 min。产物采用1%胶进行琼脂糖凝胶电泳鉴定,确定质粒是否酶切成功。用T7 High Yield RNA Transcription Kit对2 种线性质粒样品进行体外转录,并对转录成功的RNA产物采用DNase处理。下一步使用FastPure Cell/Tissue Total RNA Isolation Kit V2对RNA产物纯化,-80 ℃冰箱贮存。
重复测定5 次病毒参考样品的核酸浓度,取平均值,根据单链核糖核酸拷贝浓度计算公式,计算标准品pET-NoV GI与pET-NoV GII核酸拷贝浓度[29]。
1.3.2 引物探针设计与筛选
以MS2噬菌体(GenBank: JF719743.1)Replicase区域基因序列为靶序列,通过NCBI在线工具进行序列分析和比对,根据ERA引物和探针的设计原理,在特异性区域设计MS2噬菌体特异性ERA引物和探针(表1),并进行扩增效率分析。并以GI和GII NoV、轮状病毒、甲型肝炎病毒4 种病毒RNA作为阴性对照、ddH2O为空白对照进行RT-ERA扩增,分析其特异性,每个反应设置2 个平行,并重复实验3 次。筛选的引物探针组合详见表2,其中GI与GII NoV引物探针为实验室前期实验筛选,所有引物和探针均在北京六合华大基因科技有限公司合成。

表1 MS2噬菌体RT-ERA引物及探针Table 1 Primers and probes used for RT-ERA of MS2 phage
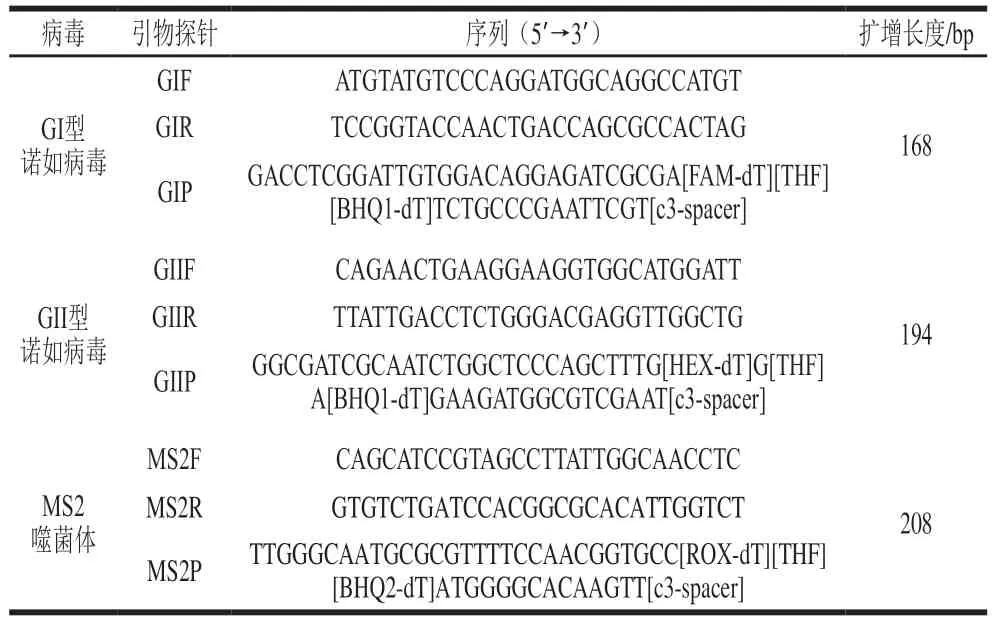
表2 GI、GII NoV与MS2噬菌体RT-ERA引物及探针序列Table 2 Primer and probe sequences used for RT-ERA of GI, GII NoV and MS2 phage
1.3.3 双重荧光RT-ERA检测体系构建
分别将GI、GII NoV与MS2噬菌体ERA引物和探针(浓度均为10 μmol/L)等体积混合,参照RT-荧光型核酸扩增试剂盒(ERA法)说明书制备每份样本的预混液(GI NoV+MS2或GII NoV+MS2):溶解剂20 μL,正向引物(GI/GII NoV+MS2)各2.1 μL,反向引物(GI/GII NoV+MS2)各2.1 μL,荧光探针(GI/GII NoV+MS2)各0.6 μL,RNA模板(浓度为106拷贝/μL)5 μL,DEPC处理过的无菌ddH2O 13.4 μL。取48 μL上述预混液转移至酶粉PCR管中溶解,振荡混匀并短暂离心。在管盖上加入2 μL的激活剂,小心盖好管盖,瞬时离心使激活剂进入预混液中,振荡混匀并再次瞬时离心,放入迷你qPCR仪中反应。
反应程序为:42 ℃、1 s;42 ℃、20 s,40 个循环。在第2反应阶段收集FAM/HEX/ROX荧光信号,其中FAM荧光(蓝色)通道检测对象为GI NoV,HEX荧光(绿色)通道检测对象为GII NoV,ROX荧光(橙色)通道检测对象为MS2过程控制病毒,同时以无菌ddH2O为空白对照,每个反应设置2 个平行,并重复实验3 次。
1.3.4 双重荧光RT-ERA扩增体系优化
1.3.4.1 引物体系优化
将上述体系中的正反向GI NoV+MS2或GII NoV+MS2混合引物(浓度均为10 μmol/L)体积分别调整至2.1、3.1、4.1、5.1 μL,其他组分不变,并相应减少ddH2O含量,使反应体系总体积保持不变,进行RT-ERA反应。
1.3.4.2 探针体系优化
采用1.3.4.1节确定的最佳引物体积,将该体系中的荧光探针(浓度均为10 μmol/L)体积分别调整至0.8、1.0、1.2、1.4、1.6、1.8 μL,其他保持组分不变,并相应减少ddH2O含量,使反应体系总体积保持不变,进行RTERA反应。
1.3.5 双重荧光RT-ERA扩增程序优化
为满足快速检测需求,在1.3.4节确定的最佳反应体系的基础上对反应程序进行优化,即反应程序第2阶段控制在42 ℃条件下,扩增程序分别调整为:15 s、40 个循环;10 s、40 个循环;5 s、40 个循环;5 s、35 个循环,分别放入迷你qPCR仪中反应,在第2反应阶段收集FAM/HEX/ROX荧光信号,同时以无菌ddH2O为空白对照,根据荧光扩增曲线结果确定最短反应时间,每个反应设置2 个平行,并重复实验3 次。
1.3.6 双重荧光RT-ERA扩增灵敏度实验
将GI NoV+MS2和GII NoV+MS2混合RNA分别进行10 倍梯度稀释,使GI和GII NoV的浓度分别为106、105、104、103、102拷贝/μL,采用1.3.4节中建立的荧光型RT-ERA快速检测程序对筛选出的最佳引物探针体积进行灵敏度分析,同时以无菌ddH2O为空白对照,每个反应设置2 个平行,并重复实验3 次。
1.3.7 真实样品中NoV RNA提取方法确定与提取效率分析
为提高贝类样品NoV RNA的提取效果,研究比较FastPure Cell/Tissue Total RNA Isolation Kit V2、PureLinkTMRNA Mini Kit、E.Z.N.A.®Viral RNA Kit、Qiagen RNeasy Mini Kit 4 种提取试剂盒对病毒RNA的提取效率。按照GB 4789.42—2016《食品微生物学检验 诺如病毒检验》[30]的规定,可通过对MS2过程控制病毒RNA的提取效率表示NoV RNA的提取效率,将10 μL MS2过程控制病毒添加到2.0 g贝类样品的消化腺中,计算4 种提取试剂盒对病毒RNA的提取效率。
首先采用MS2过程控制试剂盒制备MS2过程控制病毒RNA,并对所得RNA按照1∶3的比例使用标准物质稀释液进行梯度稀释,共稀释5 个浓度,进行实时荧光RT-qPCR扩增。以未稀释和梯度稀释的过程控制病毒的浓度为X轴(设过程控制病毒原液浓度为参照,记为1(即提取效率为100%)),以其Ct值为Y轴,绘制过程控制标准曲线。实际样本检测时MS2的Ct值在标准曲线上对应的浓度为C,则MS2过程控制病毒提取效率如下:
4 种提取试剂盒提取的RNA均使用50 μL的DEPC处理过的无菌ddH2O进行洗脱,采用智能化超微量核酸检测仪测定浓度,-80 ℃保存备用。将不同试剂盒提取的样品RNA作为扩增模板,参照MS2过程控制试剂盒说明书,进行实时荧光RT-qPCR扩增,分析提取效率选出最适合贝类样品RNA提取的试剂盒。根据GB 4789.42—2016[30],NoV提取效率/%=经病毒提取等步骤后的过程控制病毒RNA浓度×100,即Ct值对应浓度×100%。
1.3.8 双重荧光RT-ERA真实样品检测
真实样本共计29 份,其中2 份含NoV的粪便样本作为方法确证的阳性样本,27 份市售贝类作为方法适用性验证的市售样品。贝类样品参照GB 4789.42—2016[30]中NoV检测方法进行前处理,并采用1.3.7节确定的最适RNA提取的试剂盒进行提取和提取效率分析。最后以GB 4789.42—2016为参比方法,采用本研究建立的双重荧光RT-ERA方法与GB 4789.42—2016规定的荧光定量RT-qPCR方法[30]同时对29 份样品进行GI、GII NoV检测,并对2 种方法的符合性进行比较,确定本研究方法的适用性和可行性。
2 结果与分析
2.1 GI与GII NoV RNA参考样品的制备
重组质粒pET-NoV GI全长5 788 bp,质粒构建示意图见图2A。重组质粒pET-NoV GII全长5 640 bp,质粒构建示意图见图2B。测序结果表明人工合成片段正确克隆到pET28-a+载体中,证实重组质粒构建成功。使用XhoI酶对pET-NoV GI与pET-NoV GII进行线性化处理,电泳检测条带大小与预期一致(图3)。体外转录后,测定RNA平均质量浓度分别为5.73 μg/mL与5.5 μg/mL,经计算,GI NoV参考样品拷贝数为1.326×109拷贝/μL,GII NoV参考样品拷贝数为1.289×109拷贝/μL。采用GB 4789.42—2016荧光定量RT-qPCR方法[30]对RNA标准品进行GI和GII靶标成分的检测,可以获得良好的S型扩增曲线(图4),证实成功获取了GI与GII NoV RNA,可作为RNA参考样品进行后续实验。
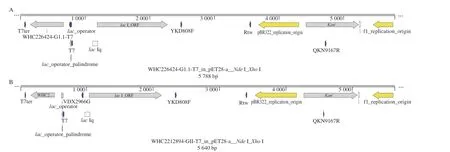
图2 重组质粒pET-NoV GI(A)与pET-NoV GII(B)构建示意图Fig.2 Schematic diagram of pET-NoV GI (A) and pET-NoV GII (B) recombinant plasmids
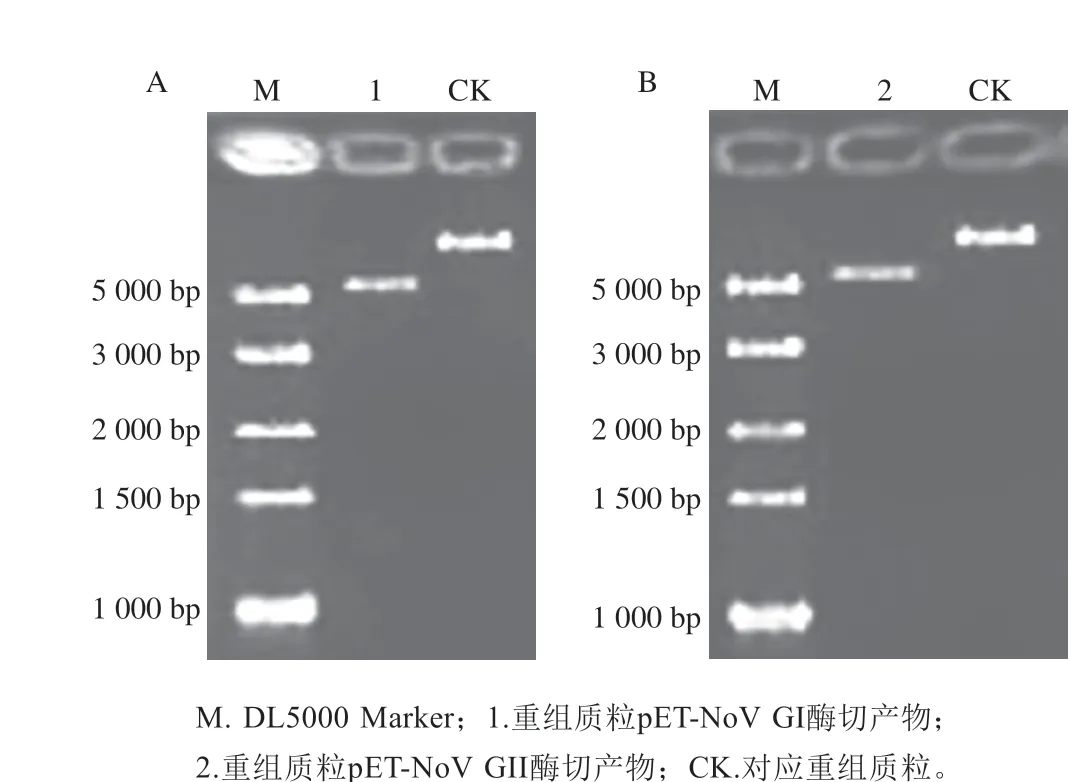
图3 pET-NoV GI(A)与pET-NoV GII(B)重组质粒酶切电泳图Fig.3 Electrophoresis of pET-NoV GI (A) and pET-NoV GII (B)recombinant plasmids
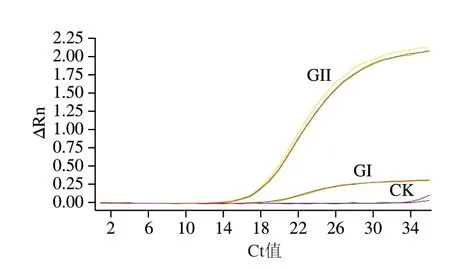
图4 GI和GII靶标基因RT-qPCR检测结果Fig.4 RT-qPCR for GI and GII target genes
2.2 NoV双重荧光RT-ERA检测方法的建立
首先筛选了MS2噬菌体ERA检测引物探针,筛选到一组MS2引物探针的扩增效果良好,特异性良好(图5A)。然后将其与前期实验室筛选的GI、GII NoV ERA引物探针分别组合,对GI、GII NoV RNA与MS2噬菌体RNA混合模板进行扩增,结果显示,GI、GII NoV均可以与MS2噬菌体组合进行双重RT-ERA荧光反应(图5B、C),但扩增效率稍低,因此需要对反应体系进行优化。
2.3 双重荧光RT-ERA扩增体系引物探针优化结果
为提高建立的双重荧光RT-ERA法的扩增效率,本研究分别对体系中引物和探针的浓度进行了优化。其中引物优化结果分别如图6A、B所示,可以看出随着引物浓度的增加荧光强度均呈逐渐增强趋势,当引物量提高到4.1 μL时,荧光强度最高,GI、GII NoV的扩增曲线与MS2噬菌体以及阴性对照能明显区分。
以4.1 μL作为最佳引物量进行探针浓度优化,结果分别如图6C、D所示,同样地,扩增曲线荧光强度也随着探针浓度的增加不断增强,但当探针量达到1.6 μL时,浓度的增加对曲线的扩增效果影响不大,说明已达到反应体系的阈值。因此,综合扩增效率与成本考虑,将1.8 μL作为最佳探针添加量。通过优化,确定双重荧光RT-ERA扩增体系中引物及探针的最佳用量分别为4.1 μL与1.8 μL。
2.4 双重荧光RT-ERA反应程序优化结果
为实现快速检测,本研究进一步对反应程序进行优化,结果如图7所示,可以看出当第2步反应程序为15 s、40 个循环时扩增效率最好(图7A),此时检测时间为17 min 10 s,相对于原始扩增程序的20 min 30 s,可以节省3 min 20 s。随着扩增时间的缩短,扩增效率虽有逐渐下降趋势(图7),但仍具有良好的扩增曲线,可以满足检测需求。其中当第2步反应程序为5 s、35 个循环时,检测时间仅需9 min 6 s(图7D),相对于原始扩增程序可以节省11 min 24 s。

图7 双重荧光RT-ERA反应时间优化结果Fig.7 Optimization of dual RT-ERA reaction time
2.5 双重荧光RT-ERA检测灵敏度分析
将10 倍梯度稀释的GI NoV+MS2和GII NoV+MS2混合RNA进行双重荧光RT-ERA检测灵敏度分析。如图8所示,建立的方法对GI与GII NoV检出限均为102拷贝/μL,具有较高的灵敏度,可以满足检测需求。

图8 双重荧光RT-ERA灵敏度分析结果Fig.8 Sensitivity of dual RT-ERA
2.6 真实样品检测结果
如图9A所示,MS2过程控制病毒RNA浓度与样品Ct值线性关系良好(R2=0.983 3),符合GB 4789.42—2016[30]规定。图9B为4 种试剂盒对真实样品中MS2 RNA的扩增效果,根据提取效率公式,PureLinkTMRNA Mini Kit对贝类样品中MS2过程控制病毒RNA的提取效率最高,约1.94%,因此后续均采用此试剂盒进行贝类样品RNA提取。

图9 不同试剂盒对贝类样品RNA的提取效率Fig.9 Extraction efficiency of RNA in shellfish samples by different kits
应用本研究建立的双重荧光RT-ERA方法与参比方法GB 4789.42—2016中的荧光RT-qPCR方法对29 份真实样品进行检测,结果显示(表3),2 份真实粪便样本可以正确检出,说明本研究建立的双重荧光RT-ERA快检方法对真实样本检测的准确性。27 份贝类样品有3 份检出GII NoV,未检出GI NoV,结果与GB 4789.42—2016一致,证实了本研究建立的方法在贝类样品中检测的适用性。
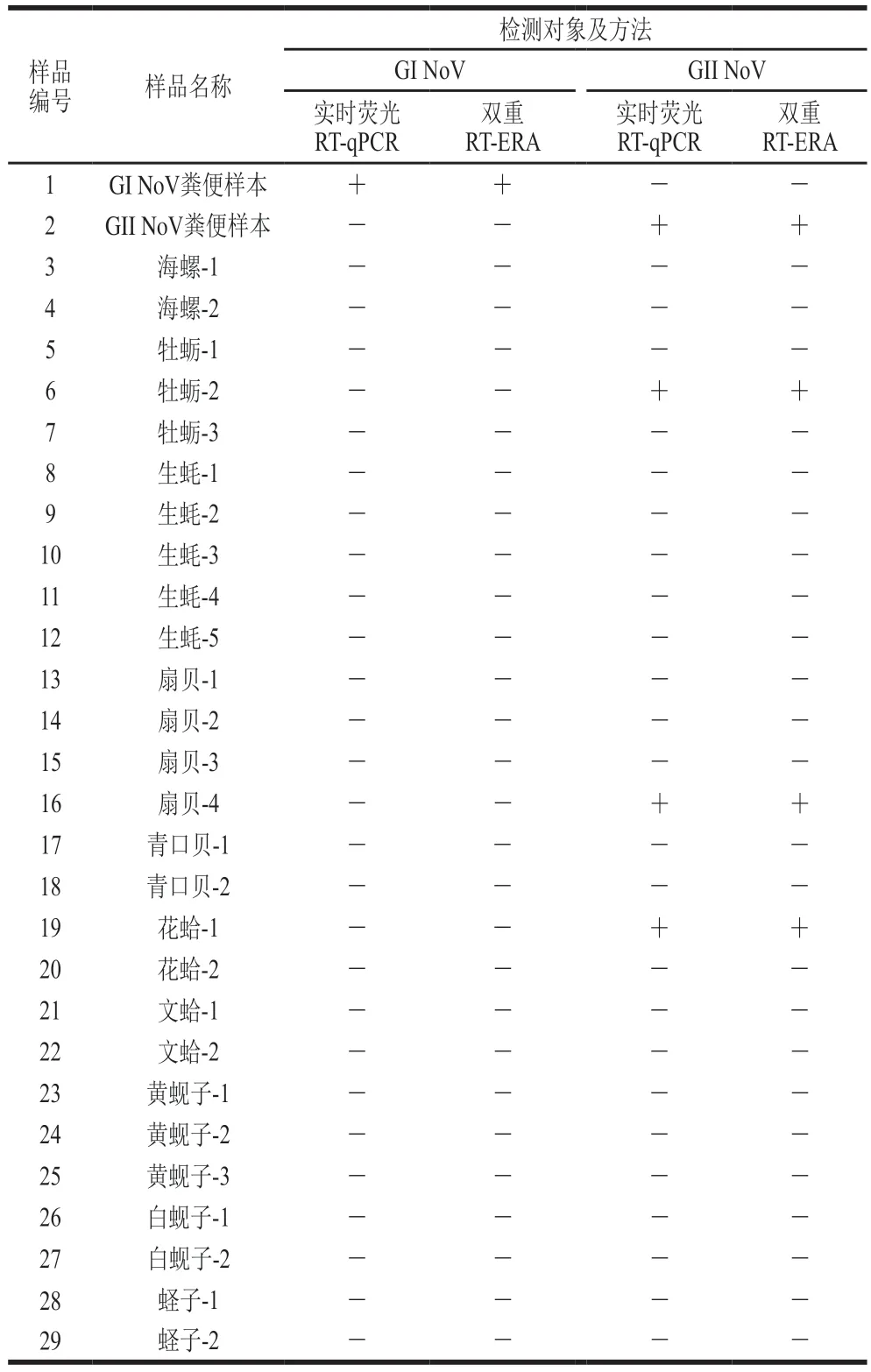
表3 真实样品检测结果Table 3 Results of detection of real samples by RT-PCR and dual TR-ERA
3 讨 论
近年来,由NoV感染引起的急性胃肠炎爆发事件已屡见不鲜,这可能与全国范围的NoV急性胃肠炎暴发监测工作起步较晚,大多数地区不具备完整的检测体系或缺乏相应的检测能力有关[31]。本研究建立的双重荧光RTERA检测方法,检测速度快、操作简单、易于推广。且可以在一个反应单元中同时检测出2 种病毒,可降低检测成本,同时也能节约检测时间。此外还通过T7 RNA聚合酶体外转录的方式获得了高纯度的RNA,研制了pET-NoV GI与pET-NoV GII两种RNA标准参考样品,可以作为检测体系中的阳性对照品,解决了因NoV无法体外培养导致的阳性样品缺乏,检测无法进行的瓶颈问题。从而为提升我国NoV早期筛查和风险防控水平,预防NoV中毒事件,保障人民身体健康提供重要的技术支撑。
本研究采用的RT-ERA技术是通过模拟生物体遗传物质自身扩增复制的原理,利用抗体制药领域先进的Molecular Design、Directed Evolution、Affinity Maturation等技术手段,将来源于细菌、病毒和噬菌体的特定工具酶进行改造突变并筛选其功能,以此建立的最优反应体系。与传统的恒温扩增技术LAMP相比,ERA技术无需设计多对引物,反应温度更低,且扩增产物可直接用于克隆测序,在灵敏度和特异性上也有了一定的提高。与目前的检测“金标准”PCR技术相比,RT-ERA技术可摆脱大型仪器的束缚,且在特异性、灵敏度良好的基础上表现出更短的检测时长。另外,RT-ERA技术的优势不仅体现在其检测时间与恒温特性上,还在于其具备现场检测的潜力,结果读取方式多种多样,如:1)与侧向流动试纸条联合,2)与显色剂结合,3)使用荧光PCR仪,均可达到可视化检测的目的。项目组前期与公司合作开发了迷你qPCR仪,机身质量为1.38 kg,尺寸为247 mm×188 mm×133 mm可同时对4 种荧光通道进行信号采集,且升降温速度快(8~10 ℃/s),无运动光学部件,移动运输无需矫正,自带人机交互和外接人机交互设计,并具有远程可控组件,可自动出具检测结果或远程出具结果,具有远程数据传输功能。本研究将研制的NoV RNA标准参考样品,建立NoV双重荧光RT-PCR技术,与迷你qPCR仪结合,以期为NoV病毒现场可视化多重快速检测和监测提供了技术与装备,对切断NoV传播,筑牢食品“防护墙”,控制NoV爆发,保障人民健康具有重要意义。
4 结 论
本研究首先通过将NoV靶序列构建到含T7启动子的载体中,通过体外转录的方式获得了高纯度的RNA,研制了pET-NoV GI与pET-NoV GII RNA标准参考样品。然后以MS2噬菌体作为过程控制病毒,建立了一种高效快速的GI与GII NoV双重荧光RT-ERA检测方法,最低可以检测出102个拷贝数量级的NoV核酸样品,检测时间最短可以缩减至10 min左右。最后将建立的方法应用于真实样品中,通过国标规定的荧光RT-qPCR比较,证实了本研究方法在真实样品中检测的准确性和适用性。同时依托合作开发的迷你qPCR仪,为NoV监测检测和现场执法提供强有力的科技支撑。
