基于BERT和CNN的基因剪接位点识别
2023-10-21左敏王虹颜文婧张青川
左敏,王虹,颜文婧,张青川*
基于BERT和CNN的基因剪接位点识别
左敏1,2,王虹1,2,颜文婧1,2,张青川1,2*
(1.北京工商大学 农产品质量安全追溯技术及应用国家工程研究中心,北京 100048; 2.北京工商大学 电商与物流学院,北京 100048)( ∗ 通信作者电子邮箱zqc1982@126.com)
随着高通量测序技术的发展,海量的基因组序列数据为了解基因组的结构提供了数据基础。剪接位点识别是基因组学研究的重要环节,在基因发现和确定基因结构方面发挥着重要作用,且有利于理解基因性状的表达。针对现有模型对脱氧核糖核酸(DNA)序列高维特征提取能力不足的问题,构建了由BERT(Bidirectional Encoder Representations from Transformer)和平行的卷积神经网络(CNN)组合而成的剪接位点预测模型——BERT-splice。首先,采用BERT预训练方法训练DNA语言模型,从而提取DNA序列的上下文动态关联特征,并且使用高维矩阵映射DNA序列特征;其次,采用人类参考基因组序列hg19数据,使用DNA语言模型将该数据映射为高维矩阵后作为平行CNN分类器的输入进行再训练;最后,在上述基础上构建了剪接位点预测模型。实验结果表明,BERT-splice模型在DNA剪接位点供体集上的预测准确率为96.55%,在受体集上的准确率为95.80%,相较于BERT与循环卷积神经网络(RCNN)构建的预测模型BERT-RCNN分别提高了1.55%和1.72%;同时,在5条完整的人类基因序列上测试得到的所提模型的供体/受体剪接位点平均假阳性率(FPR)为4.74%。以上验证了BERT-splice模型用于基因剪接位点预测的有效性。
剪接位点识别;BERT;卷积神经网络;深度学习;脱氧核糖核酸
0 引言
近年,高通量测序技术产生了海量的基因组序列,在增加生物序列数量的同时,扩大了对自动化序列分析计算技术的需求,给基因结构注释领域的研究带来了机遇和挑战[1]。剪接位点识别是基因结构注释研究领域的重要方向。在真核生物中,一个基因由多个外显子和内含子组成,外显子与内含子的边界称为剪接位点,基因剪接是基因表达中的重要过程,可以影响蛋白质翻译的结果和人体生命活动。因此,正确识别剪接位点可以更好地了解基因表达过程,在生物医学研究中发挥重要作用。
一个脱氧核糖核酸(DeoxyriboNucleic Acid, DNA)分子可以看作是由A、C、G和T这4个字组成的序列,分别代表腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶这4种碱基。剪接位点的结构规则遵从“GT-AG”规则,即外显子到内含子的边界称为供体剪接位点,通常表现为一个保守的二核苷酸——鸟嘌呤和胸腺嘧啶(Guanine and Thymine, GT);内含子到外显子的边界称为受体剪接位点,通常表现为一个保守的二核苷酸——腺嘌呤和鸟嘌呤(Adenine and Guanine, AG),如图1所示。

图1 剪接位点示意图
传统的生物检测方法检测DNA剪接位点的成本高、耗时长,而机器学习方法识别剪接位点是一种更经济、有效的解决方案。一条DNA序列包含大量GT/AG,因此供体/受体剪接位点的识别通常被转化为两个不同的二元分类问题,即区分一条DNA序列是否包含真剪接位点。近年,在DNA结构注释研究领域广泛使用的机器学习方法包括支持向量机(Support Vector Machine, SVM)[2-4]、马尔可夫模型(Markov Model, MM)[5-6]、随机森林(Random Forest, RF)[7-8]、贝叶斯网络(Bayesian Network, BN)[9]和条件随机场(Conditional Random Field, CRF)[10]等,均取得了良好效果。最近,相关领域引入了深度学习方法可以基于大量数据集的训练自动提取最相关的特征,解决了机器学习方法需要手动定义特征集、捕捉DNA序列高维特征难的缺点,呈现很强的端到端预测能力[11]。大多数深度学习方法基于卷积神经网络(Convolutional Neural Network, CNN),如DSSP(Deep Splice Site Prediction system)[12]、SpliceRover[13]、SpliceFinder[14]、SpliceAI[15]、Spliceator[16]和Deep Splicer[17],预测剪接位点;其他一些工具[18]专注于DNA的序列特征,采用基于循环神经网络(Recurrent Neural Network, RNN)模型,如长短期记忆(Long Short-Term Memory, LSTM)和门控循环单位(Gated Recurrent Unit, GRU),捕捉状态之间的依赖性。虽然这些工具在剪接位点预测取得良好的效果,但是CNN提取局部特征的能力受滤波器大小的限制,通常无法捕获上下文中的语义依赖关系。RNN模型(如LSTM、GRU)虽然具有学习长期依赖的能力,但当输入序列较长时,由于对过去所有的状态顺序处理,且压缩上下文信息,存在梯度消失和效率低的问题。因此需要一种既能够提取局部特征,又能够全面考虑所有上下文信息的方法,从而更好地模拟DNA序列。
此外,深度学习方法依赖于相关研究问题的高质量数据集,在剪接位点的预测研究中,大多采用HS3D数据集[19]。该数据集的剪接位点位于中间,样本长度为140核苷酸(nucleotide, nt),后续研究[20-22]大都沿用了该数据集。近期,研究人员探究了剪接位点预测任务所使用数据集的最佳输入长度,如SpliceFinder测试了40~400 nt的序列长度,发现在扩大负样本集前,长度对准确率的影响较小,扩大负样本集后,更长的序列有助于模型保持良好的性能,研究任务最终选择400 nt作为较优输入长度。Spliceator测试了从20~600 nt长度的数据集,发现序列长度增加至200 nt后,预测精度较高。Deep Splicer将神经网络的输入长度分别设置为261、401、1 001和2 001,发现401 nt和1 001 nt作为输入训练的模型比其他模型假阳性更少,但考虑到精度和计算成本,最终认为401 nt长度可以在计算成本和准确率中取得平衡。
基于上述讨论,本文设计了由BERT(Bidirectional Encoder Representations from Transformer)和平行CNN组合而成的剪接位点预测模型——BERT-splice;同时,基于模型的学习机制观测,为剪接位点预测任务的最优输入序列长度提供指导。通过集合DNA序列特性与深度学习技术,解决了DNA序列特征的表示和提取问题,实现了对供体/受体剪接位点的准确预测。
1 BERT⁃splice模型
基于DNA序列、蛋白质序列等生物序列与文本信息的相似之处,一些研究人员在DNA注释研究领域引入自然语言处理技术,以文本的形式从生物数据中学习有用的特征,并取得了较好的效果[23-25]。BERT-splice模型是一种基于预训练DNA语言模型编码的混合模型架构。首先,使用BERT层通过查询字向量表将DNA序列中的每个核苷酸转换为一维向量,作为模型输入;其次,提取BERT层中最后一层Transformer的输出作为CNN层的输入;最后,将剪接位点的检测问题转换为二分类问题。分类由全连接层(Fully Connected layer, FC)实施,该层将BERT字向量和多个平行CNN提取的特征映射为输出。使用Sigmoid函数输出预测概率,以确定是否是剪接位点。BERT-splice模型的框架见图2。

图2 BERT-splice模型框架
1.1 DNA预训练模型
本文使用BERT预训练提取DNA序列的高维“语义”特征,BERT通过自我监督的方式使用未标记数据学习DNA的基本“语法”和“语义”,并促进下游任务的继续训练。BERT由12个相同的Transformer编码块串接,每个Transformer编码块由一个多头自注意机制和一个全连接的前馈神经网络构成,如图3所示。
注意力函数Attention可以描述为值向量的加权和,其中分配给每个值向量的权重通过查询向量与相应键向量的相似性函数计算,计算公式如式(1)所示:
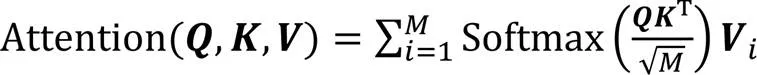
其中M为K的维度。
多头注意机制执行不同的可学习线性投影,将、和集合平行地转换为个子空间。独立注意力输出被连接并再次投射到集合的同一个维度,从而产生多头注意值:

自注意力层的输出在进入前馈神经网络之前会经过残差连接与归一化层,归一化之后的文本向量送入前馈神经网络,它主要包含一个线性变换和一个采用ReLU(Rectified Linear Unit)激活函数的非线性变换两层结构,如式(4)所示:

其中:为前馈神经网络的输入;1、2为权重向量;1、2为偏置。
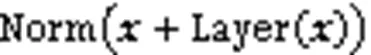
1.2 剪接位点预测模型
本文构建了一个基于预训练BERT模型所形成的DNA语义模型,结合多个平行CNN的分类器识别剪接位点,如图4所示。

图4 平行CNN结构
由于DNA序列中有意义的片段(motif)没有明确的长度,因此采用多尺度的特征有助于模型学习。本文使用不同滤波尺度的平行CNN结构,提取DNA序列中的特征,再拼接多尺度特征,实现基于多尺度特征的有效识别。首先序列输入基于BERT预训练的DNA语言模型,获得合理的初始值作为CNN的输入,使得继续训练的CNN分类器可以在样本数有限的情况下快速收敛,同时微调BERT模型。BERT的结构与DNA预训练模型相同(见1.1节),多个平行的CNN包括输入层、卷积层、池化层、ReLU层和全连接层。本文设置了6个平行的多尺度卷积层,每层有256个滤波器,卷积层的维度分别设置为3、4、5、9、10和11。
1.3 损失函数
本文使用二值交叉熵(Binary Cross Entropy)计算预测结果x和真实结果y的损失。计算公式如下:

2 实验过程
2.1 实验数据集
本文实验使用了独立构建的数据集。参考基因组hg19数据集(FASTA文件)和相应序列的注释(BED文件)(从UCSC下载),最终构造了用于训练DNA语言模型的数据集1和用于训练剪接位点预测模型的数据集2。
数据集1将hg19数据集随机切分为512 nt的长度,最终生成164万条序列用于预训练DNA语言模型。
数据集2由两个子集组成,分别是供体数据集(Donor Splice Sites, DSS)、受体数据集(Acceptor Splice Sites, ASS)。由于剪接位点相邻的核苷酸在剪接机制中发挥着重要作用,为了减少因序列长度导致剪接位点附近外显子/内含子信息的缺失,在构造数据集时将剪接位点周围外显子/内含子相同长度的基因片段包含在内,即保证剪接位点上下游序列长度相等。本文以剪接位点二核苷酸为中心截取特定长度的基因片段,去除非标准剪接位点,删除重叠样本,作为正样本数据集。正样本序列的构建将剪接位点位于序列中间位置,并保持内含子与外显子序列长度均等。正样本数据集最终包含供体剪接位点数为74 192,受体剪接位点数为80 704。负样本选取基因区域与正样本集非重叠部分,随机截取与正样本相同的长度,保证二核苷酸GT/AG与供体/受体剪接位点位于相同的位置,负样本数与正样本数相等。最终将数据集按8∶1∶1划分为训练集、验证集和独立测试集,如表1所示。为了研究不同输入长度对模型的影响,本文通过改变截取外显子/内含子长度25~200 nt作为输入,即模型输入长度为50~400 nt,如图5所示。

表1 数据集2的统计信息

图5 选择不同长度的DNA序列
2.2 BERT预训练
本文将每一个核苷酸看作一个字,将数据集1按照单个碱基进行分词,作为预训练DNA语言模型的输入。
本文采用掩码语言模型(Masked Language Model, MLM)方法,该模型简单随机地将一部分输入替换为掩码标记,通过预测这些掩码标记实现训练策略(如图6所示)。本文将语料库中15%的区域随机替换为掩码令牌,其中这些掩码令牌中,只有80%被真正替换为掩码标记,10%未被替换,为了防止预测中先验信息的泄漏将剩余10%替换为随机信息。

图6 采用MLM方法训练BERT
2.3 超参数设置
本文的实验环境如下:操作系统为Windows10、64 bit,处理器为Intel Core i7-1165G7 CPU,显卡为NVIDIA GeForce RTX3060,显存为12 GB。编程环境为Python3.9,PyTorch1.9.1。模型参数设置如表2所示。

表2 模型参数设置
在模型训练过程中还使用了“提前停止(Early Stopping)”技巧,即当验证集上的损失不再下降时及时停止训练,以此避免过拟合、不收敛等,并提高模型训练效率。
2.4 评价指标
本文利用准确率(accuracy)、敏感性 (Sensitivity, Sn) 、特异性 (Specificity, Sp)、假阳性率(False Positive Rate, FPR)、Matthew相关系数(Matthews Correlation Coefficient, MCC)、ROC(Receiver Operating Characteristic)曲线下面积(Area Under Curve, AUC)和Top-准确率(Top-acc)评估模型性能。计算公式如下:





其中:真阳性表示正确预测剪接位点的数量;真阴性表示正确预测非剪接位点的数量;假阳性表示将非剪接位点预测为剪接位点的数量;假阴性表示将剪接位点预测为非剪接位点的数量。
MCC考虑、、和检查二元分类的质量,+1表示完美预测,0表示平均随机预测,-1表示逆预测。
ROC曲线表示真阳性率(True Positive Rate, TPR)与假阳性率的关系。它描述了真阳性和假阳性之间的相对权衡,可以在整个类分布范围内比较分类器的性能。AUC计算ROC下的区域。如果AUC接近0.5,则性能接近随机;如果AUC接近1,则性能接近完美。
因为DNA序列中的大多数位置不是剪接位点,所以也评估了Top-准确率:假设在基因组序列中,有个位置是供体或受体位点,在使用BERT-splice预测基因中每个二核苷酸GT/AG的类别后,降序排列它的预测概率,从预测概率的有序列表中选择前个核苷酸位置,这个核苷酸中正确分类的核苷酸的比例被称为Top-准确率。本文计算了Top-50%准确率,即计算二核苷酸列表中前50%二核苷酸内正确分类的比例。
3 实验与结果分析
3.1 测试不同长度的输入
为了选择最适合训练的区域,本文使用50~400 nt长度的序列作为模型的输入,如表3所示。在独立测试集上,对于供体位点,所有长度的平均准确率为96.40%。当长度为300 nt时,准确率最高为96.88%。对于受体剪接位点,相较于供体剪接位点平均准确率有所下降,为95.24%。当序列长度为300 nt时,准确率为95.80%。由此得出,更长的序列有助于模型保持良好的性能。因此在接下的实验中,本文使用300 nt的序列长度预测供体和受体位点。
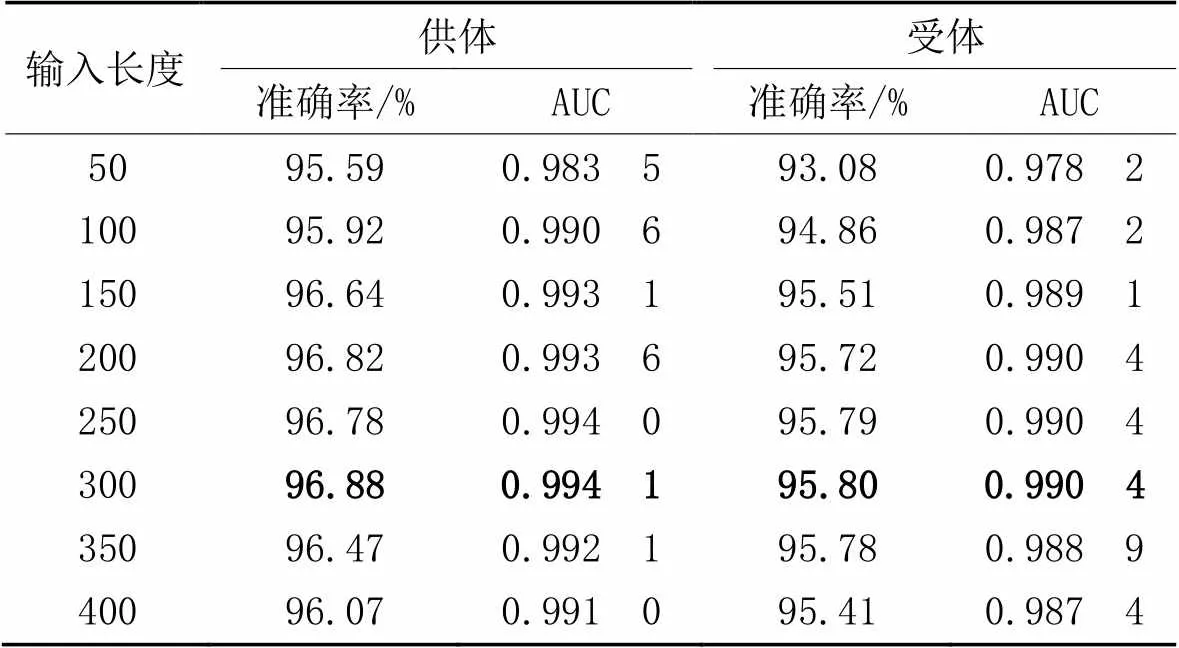
表3 不同输入长度的序列在独立测试集上的准确率和AUC
3.2 不同特征提取模型的性能比较
本文使用BERT模型、Word2Vec[26]和fastText[27]生成序列特征嵌入信息。为了对性能进行公平的比较,将预先训练好的特征嵌入模型与相同结构的平行CNN进行连接。特征嵌入模型的预训练均使用数据集1,剪接位点预测任务使用数据集2,在独立测试集上性能评估的结果如表4所示。
如表4所示,基于BERT特征嵌入的预测器获得了最好的性能,表明BERT生成的嵌入信息可以提供最有效的DNA理解,有利于促进下游分类器获得更好的效果。

表4 不同模型在独立测试集上的性能比较
3.3 不同分类模型的性能比较
为了评估BERT-splice的预测性能,本文选取了不同的分类器替换平行CNN作为对比基线算法,包括BERT、BERT与双向长短期记忆(Bi-directional Long Short-Term Memory, BiLSTM)网络结合的BERT-BiLSTM[28]、BERT与循环卷积神经网络(Recurrent Convolutional Neural Network, RCNN)结合的BERT-RCNN[29]。
不同模型在数据集2独立测试集上的预测结果如表5所示。可以看出,本文提出的BERT-splice无论在供体剪接位点还是受体剪接位点上的测试结果均为最优,在DNA剪接位点供体集上的预测准确率为96.55%,在受体集上的准确率为95.80%,相较于BERT-RCNN分别提高了1.55%和1.72%。BERT-splice模型的分类效果优于对比模型。结果充分说明,BERT-splice能够对DNA序列特征进行更高效的利用,能够提升预测效果。
3.4 人类基因序列测试
本节通过预测人类基因序列上的剪接位点进一步验证模型的泛化性能,所采用的验证基因,没有在模型训练和继续训练过程中使用,所选择基因序列上供体/受体剪接位点数不少于4个。由于基因序列含有大量的GT/AG二核苷酸位点,对于测试的每一条基因,正负样本数高度不平衡(如表6所示)。通过以GT/AG为中心的滑动窗口,选取300 nt长度的序列作为模型输入,预测每一个GT/AG位点,判断滑动窗口中心的二核苷酸是供体或是其他通用核苷酸/受体,或是其他通用核苷酸。一旦预测了每个序列,则利用预测概率将供体和受体的预测位点按降序排列。评估Top-50%准确率、模型预测的假阳性,实验结果如表6所示。可以看出,供体和受体剪接位点平均Top-50%准确率为95.37%,平均假阳性率为4.74%。总体上,BERT-splice适用于预测基因序列剪接位点预测。
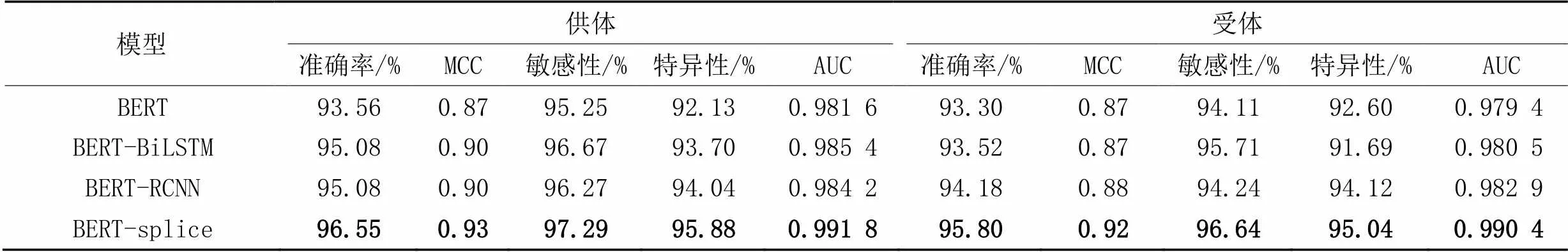
表5 本文模型与常用的分类模型在独立测试集上的性能比较
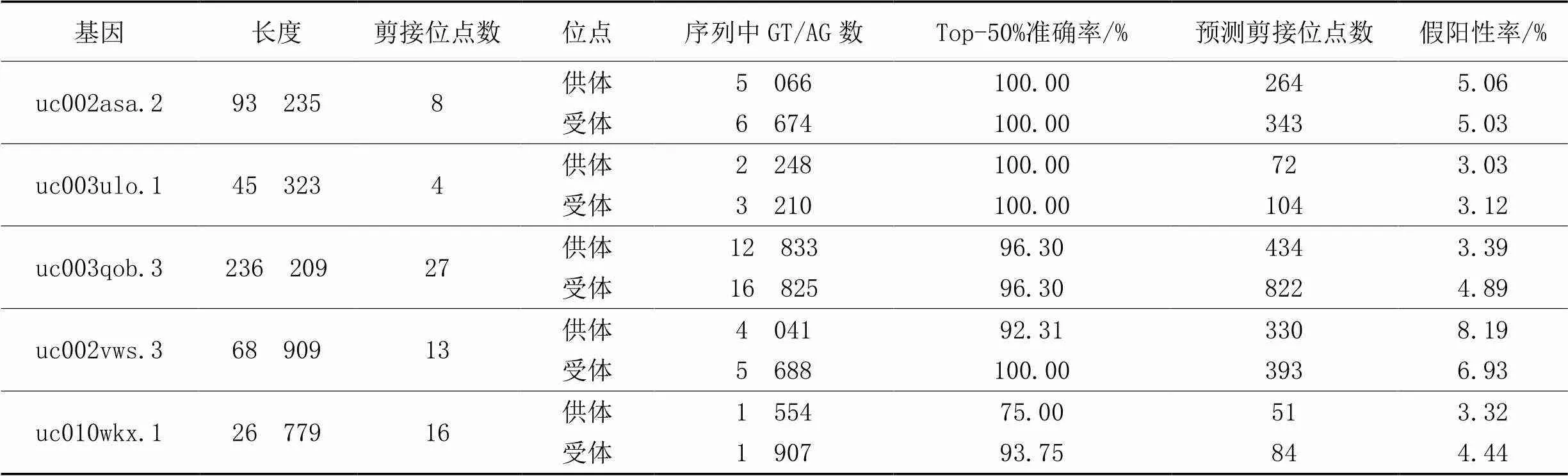
表6 基于BERT-splice模型的人类基因预测结果
4 结语
剪接位点的识别在真核生物基因结构预测中至关重要,本文提出了一种剪接位点预测模型BERT-splice。BERT-splice模型使用BERT的预训练对DNA序列进行高维特征体系表示,实现对人类基因组序列上下文动态关联特征的有效嵌入;同时,集成多个平行CNN,进一步提取局部相关特征,实现了对DNA剪接位点的准确识别;最后,基于深度学习的决策机制,为剪接位点预测任务的最优输入长度提供指导。实验结果表明,基于预训练语言模型的BERT-splice提取的向量表示在应用上效果优于Word2Vec和fastText的特征向量;同时,与其他分类模型(BERT-BiLSTM和BERT-RCNN)相比,多个平行的CNN在处理剪接位点识别的任务上性能最优。本文基于多种序列的长度测试,认为长度为300 nt的序列可以涵盖所需决策信息,在准确率和计算成本消耗上达到一定平衡。最后,本文预测了完整的基因序列,取得了良好的效果。在剪接位点预测模型成功构建的基础上,下一步工作将涉及将该模型应用于其他物种和其他生物元件的识别。这是为了扩大模型的适用范围,提供准确的预测和识别工具,为相关研究提供更全面的支持。
[1] WAINBERG M, MERICO D, DELONG A, et al. Deep learning in biomedicine[J]. Nature Biotechnology, 2018, 36(9): 829-838.
[2] DEGROEVE S, SAEYS Y, DE BAETS B, et al. SpliceMachine: predicting splice sites from high-dimensional local context representations[J]. Bioinformatics, 2005, 21(8):1332-1338.
[3] SONNENBURG S O R, SCHWEIKERT G, PHILIPS P, et al. Accurate splice site prediction using support vector machines[J]. BMC Bioinformatics, 2007, 8(S10): No.S7.
[4] MAJI S, GARG D. Hybrid approach using SVM and MM2 in splice site junction identification[J]. Current Bioinformatics, 2014, 9(1): 76-85.
[5] PASHAEI E, YILMAZ A, OZEN M, et al. A novel method for splice sites prediction using sequence component and hidden Markov model[C]// Proceedings of the 38th Annual International Conference of the IEEE Engineering in Medicine and Biology Society. Piscataway: IEEE, 2016: 3076-3079.
[6] ZHANG Q, PENG Q, ZHANG Q, et al. Splice sites prediction of Human genome using length-variable Markov model and feature selection[J]. Expert Systems with Applications, 2010, 37(4): 2771-2782.
[7] PASHAEI E, OZEN M, AYDIN N. Splice site identification in human genome using random forest[J]. Health and Technology, 2017, 7(1): 141-152.
[8] MEHER P K, SAHU T K, RAO A R. Prediction of donor splice sites using random forest with a new sequence encoding approach[J]. BioData Mining, 2016, 9: No.4.
[9] CHEN T M, LU C C, LI W H. Prediction of splice sites with dependency graphs and their expanded bayesian networks[J]. Bioinformatics, 2005, 21(4): 471-482.
[10] SUN S, DONG Z, ZHAO J. Conditional random fields for multiview sequential data modeling[J]. IEEE Transactions on Neural Networks and Learning Systems, 2022, 33(3): 1242-1253.
[11] LeCUN Y, BENGIO Y, HINTON G. Deep learning[J]. Nature, 2015, 521(7553): 436-444.
[12] NAITO T. Human splice-site prediction with deep neural networks[J]. Journal of Computational Biology, 2018, 25(8): 954-961.
[13] ZUALLAERT J, GODIN F, KIM M, et al. SpliceRover: interpretable convolutional neural networks for improved splice site prediction[J]. Bioinformatics, 2018, 34(24): 4180-4188.
[14] WANG R, WANG Z, WANG J, et al. SpliceFinder: ab initio prediction of splice sites using convolutional neural network[J]. BMC Bioinformatics, 2019, 20(S23): No.652.
[15] JAGANATHAN K, PANAGIOTOPOULOU S K, McRAE J F, et al. Predicting splicing from primary sequence with deep learning[J]. Cell, 2019, 176(3): 535-548.e24.
[16] SCALZITTI N, KRESS A, ORHAND R, et al. Spliceator: multi-species splice site prediction using convolutional neural networks[J]. BMC Bioinformatics, 2021, 22: No.561.
[17] FERNANDEZ-CASTILLO E, BARBOSA-SANTILLÁN L I, FALCON-MORALES L, et al. Deep Splicer: a CNN model for splice site prediction in genetic sequences[J]. Genes, 2022, 13(5): No.907.
[18] CANATALAY P J, UCAN O N. A bidirectional LSTM-RNN and GRU method to exon prediction using splice-site mapping[J]. Applied Sciences, 2022, 12(9): No.4390.
[19] POLLASTRO P, RAMPONE S. HS3D, a dataset of Homo Sapiens Splice regions, and its extraction procedure from a major public database[J]. International Journal of Modern Physics C, 2002, 13(8): 1105-1117.
[20] TAYARA H, TAHIR M, CHONG K T. iSS-CNN: identifying splicing sites using convolution neural network[J]. Chemometrics and Intelligent Laboratory Systems, 2019, 188: 63-69.
[21] DASARI C M, BHUKYA R. InterSSPP: investigating patterns through interpretable deep neural networks for accurate splice signal prediction[J]. Chemometrics and Intelligent Laboratory Systems, 2020, 206: No.104144.
[22] DU X, YAO Y, DIAO Y, et al. DeepSS: exploring splice site motif through convolutional neural network directly from DNA sequence[J]. IEEE Access, 2018, 6: 32958-32978.
[23] DO D T, LE T Q T, LE N Q K. Using deep neural networks and biological subwords to detect protein S-sulfenylation sites[J]. Briefings in Bioinformatics, 2021, 22(3): No.bbaa128.
[24] HAMID M N, FRIEDBERG I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks[J]. Bioinformatics, 2019, 35(12): 2009-2016.
[25] 张海丰,曾诚,潘列,等. 结合BERT和特征投影网络的新闻主题文本分类方法[J]. 计算机应用, 2022, 42(4): 1116-1124.(ZHANG H F, ZENG C, PAN L, et al. News topic text classification method based on BERT and feature projection network[J]. Journal of Computer Applications, 2022, 42(4): 1116-1124.)
[26] ASGARI E, MOFRAD M R K. Continuous distributed representation of biological sequences for deep proteomics and genomics[J]. PLoS ONE, 2015, 10(11): No.e0141287.
[27] JOULIN A, GRAVE E, BOJANOWSKI P, et al. Bag of tricks for efficient text classification[C]// Proceedings of the 15th Conference of the European Chapter of the Association for Computational Linguistics: Volume 2, Short Papers. Stroudsburg, PA: ACL, 2017: 427-431.
[28] GRAVES A, JAITLY N, MOHAMED A R. Hybrid speech recognition with deep bidirectional LSTM[C]// Proceedings of the 2013 IEEE Workshop on Automatic Speech Recognition and Understanding. Piscataway: IEEE, 2013: 273-278.
[29] LAI S, XU L, LIU K, et al. Recurrent convolutional neural networks for text classification[C]// Proceedings of the 29th AAAI Conference on Artificial Intelligence. Palo Alto, CA: AAAI Press, 2015: 2267-2273.
Gene splice site identification based on BERT and CNN
ZUO Min1,2, WANG Hong1,2, YAN Wenjing1,2, ZHANG Qingchuan1,2*
(1⁃,,100048,;2⁃,,100048,)
With the development of high-throughput sequencing technology, massive genome sequence data provide a data basis to understand the structure of genome. As an essential part of genomics research, splice site identification plays a vital role in gene discovery and determination of gene structure, and is of great importance for understanding the expression of gene traits. To address the problem that existing models cannot extract high-dimensional features of DNA (DeoxyriboNucleic Acid) sequences sufficiently, a splice site prediction model consisted of BERT (Bidirectional Encoder Representations from Transformers) and parallel Convolutional Neural Network (CNN) was constructed, namely BERT-splice. Firstly, the DNA language model was trained by BERT pre-training method to extract the contextual dynamic association features of DNA sequences and map DNA sequence features with a high-dimensional matrix. Then, the DNA language model was used to map the human reference genome sequence hg19 data into a high-dimensional matrix, and the result was adopted as input of parallel CNN classifier for retraining. Finally, a splice site prediction model was constructed on the basis of the above. Experimental results show that the prediction accuracy of BERT-splice model is 96.55% on the donor set of DNA splice sites and 95.80% on the acceptor set, which improved by 1.55% and 1.72% respectively, compared to that of the BERT and Recurrent Convolutional Neural Network (RCNN) constructed prediction model BERT-RCNN. Meanwhile, the average False Positive Rate (FPR) of donor/acceptor splice sites tested on five complete human gene sequences is 4.74%. The above verifies that the effectiveness of BERT-splice model for gene splice site prediction.
splice site identification; Bidirectional Encoder Representations from Transformers (BERT); Convolutional Neural Network (CNN); deep learning; DeoxyriboNucleic Acid (DNA)
This work is partially supported by National Natural Science Foundation of China (61873027).
ZUO Min,born in 1973, Ph. D., professor. His research interests include food big data, deep learning.
WANG Hong, born in 1997, M. S. candidate. Her research interests include natural language processing.
YAN Wenjing, born in 1985, Ph. D., lecturer. Her research interests include intelligent processing of biological information, deep learning, image recognition.
ZHANG Qingchuan, born in 1982, Ph. D., associate professor. His research interests include natural language processing, deep learning, information extraction.
1001-9081(2023)10-3309-06
10.11772/j.issn.1001-9081.2022091447
2022⁃09⁃29;
2022⁃12⁃22;
国家自然科学基金项目资助项目(61873027)。
左敏(1973—),男,安徽铜陵人,教授,博士,主要研究方向:食品大数据、深度学习; 王虹(1997—),女,山西大同人,硕士研究生,主要研究方向:自然语言处理; 颜文婧(1985—),女,安徽淮南人,讲师,博士,主要研究方向:生物信息智能处理、深度学习、图像识别; 张青川(1982—),男,河北石家庄人,副教授,博士,主要研究方向:自然语言处理、深度学习、信息抽取。
TP399
A
2023⁃01⁃03。
