江西新岗山森林昆虫种内遗传距离研究
2023-10-20周青松曹焕喜陈婧婷王明强谢婷婷杨娟娟陶双伦罗阿蓉朱朝东
马 奉,周青松,曹焕喜,陈婧婷,4,王明强,5,谢婷婷,杨娟娟,4,陶双伦,张 峰,罗阿蓉*,朱朝东,4
(1. 吉首大学生物资源与环境科学学院,湖南吉首 416000;2. 中国科学院动物研究所动物进化与系统学(院)重点实验室,北京 100101;3. 中国科学院动物研究所国家动物标本资源库,北京 100101;4. 中国科学院大学生命科学学院,北京 100049;5. 中国科学院成都生物 研究所山地生态恢复与生物资源利用重点实验室与生态恢复生物多样性保护四川省重点实验室,成都 610041;6. 南京农业大学植物保护学院,南京 210095)
昆虫种类繁多,生物学特性复杂多样,在生态系统中发挥着植食、分解、传粉、寄生、捕食等服务功能,是生物多样性的重要组成部分,在生态系统中占有重要地位(彩万志等, 2011;Kunin, 2019;王明强等, 2022)。准确、快速地进行物种鉴定是昆虫生物多样性监测评估等研究的基础(Dayrat, 2005)。在DNA分类学出现以前,昆虫分类学家在发现和描述物种时通常基于形态学特征,不仅耗时、费力,而且需要长时间形态学知识和经验的积累(Hongetal., 2022; Zhuetal., 2022)。因此,面对大量昆虫物种鉴定需求,形态分类学在描述昆虫物种多样性时往往很难提供高效、及时的支撑(Hebertetal., 2003; Yuetal., 2012)。此外,雌雄二型、变态发育、表型可塑与遗传可变性等问题,还会导致模棱两可甚至相互矛盾的鉴定结果(Knowlton, 1993; Jarman and Elliott, 2000; Hebertetal., 2003)。因此,加强对多类群和大样本量物种界定方法的探索,提升昆虫分类学效率,在昆虫多样性研究工作中显得非常重要而紧迫。
本世纪初以来,DNA条形码(DNA barcoding)技术为分类学研究提供了新的技术支撑,促使昆虫物种鉴定和多样性监测方法更加经济、高效,极大地提升了对已知物种、研究尚不充分或有待发现类群的物种界定效力(Hebertetal., 2003)。该技术利用标准化的基因片段作为分子标记,基于种内遗传距离小于种间遗传距离的原则,在很多类群中实现物种识别及鉴定(Hebertetal., 2003; Hebert and Gregory, 2005)。选取合适的分子标记是使用DNA条形码开展物种鉴定的关键因素(Waugh, 2007; Luoetal., 2011)。相较于核基因,线粒体基因具有母系遗传、进化速率快、基因重组率发生率低等特点(刘青青和董志军, 2018)。其中,线粒体细胞色素C氧化酶亚基I基因(Cytochromecoxidase subunit I, 简称COI)不存在内含子,在保证足够变异的情况下,容易被通用引物扩增,被普遍用作动物各类群的通用条形码区域(Hebertetal., 2003)。目前,基于COI基因的DNA条形码在昆虫物种鉴定、隐存种发现及种间互作关系等研究中已得到广泛应用和验证。例如:Hebert等(2004a)利用DNA条形码对哥斯达黎加鳞翅目弄蝶科昆虫Astraptesfulgerator的研究,发现了隐存种;Pentinsaari等(2014)对北欧1 872个甲虫的COI条形码区域进行了测序和有效识别,加速了甲虫新物种的发现;Wang等(2020, 2021)利用COI基因对大量鳞翅目幼虫进行了物种界定,进一步阐明了植食性昆虫与植物之间的互作关系。此外,部分学者甚至单独应用DNA条形码开展昆虫分类研究(Meierottoetal., 2019; Sharkeyetal., 2021)。
随着基因组测序技术的发展,DNA宏条形码(DNA metabarcoding)作为DNA条形码技术的拓展,已经在一定程度上克服了物种多样性研究中的取样瓶颈,能解决易获得但形态较难区分的昆虫及土壤、水体等环境样品分析中的问题(Thomsen and Willerslev, 2015; Deineretal., 2017; 高养春等, 2020)。不过,尽管新的技术在测序通量和效率等方面有了大幅提升,DNA宏条形码在数据产生过程中容易受PCR扩增偏差、测序深度等因素的影响;其后续的数据处理需要较复杂的生物信息学分析;所获数据在物种注释过程中能鉴定到的物种水平也依赖于物种数据库的建立和完整性(Yuetal., 2012; 王萌等, 2021)。因此,DNA宏条形码所恢复的生物多样性信息,仍然可能与实际情况存在较大偏差。相比之下,DNA条形码在物种鉴定准确性、高效性等方面依然具有综合优势,在昆虫物种鉴定和生物多样性研究中仍占据重要地位(Zahirietal., 2017; DeSalle and Goldstein, 2019; Wangetal., 2020; Wangetal., 2021)。
长期以来,昆虫DNA条形码研究工作大多局限于某个具体类群,因此其对不同昆虫类群物种多样性评估的功效一直存在争议。例如,Meier等(2006)利用1 333条COI序列,对449种双翅目昆虫进行了分子分类学研究,获得了较低的物种鉴定成功率(<70%);不过,Ashfaq等(2018)在利用DNA条形码开展多样性调查中,则发现双翅目具有较高的物种分辨率(92%)。又如:Hebert等(2003)曾提出鳞翅目昆虫的物种区分阈值为3%;蜉蝣目、毛翅目的有效阈值却为2% (Zhouetal., 2010; Webbetal., 2012)。本研究对来自中国江西新岗山的昆虫样品开展DNA条形码研究,比较不同类群所获MOTU(Molecular operational taxonomic unit)的种内遗传距离,期望进一步阐释DNA条形码在不同昆虫类群的作用功效。
1 材料与方法
1.1 研究地点和样本
采样地点位于我国江西省新岗山,其为中国亚热带森林生物多样性与生态系统功能实验样地(Biodiversity-ecosystem functioning experiment China, 简称BEF-China)(29°08′~29°11′N、117°90′~117°93′E)(马克平, 2013);其地处亚热带,年平均气温16.7℃,年均降雨量1 821 mm,具典型的季风气候(Yangetal., 2013; Bruelheideetal., 2014)。
本研究于2020年利用马来氏网在BEF-China样地中采集昆虫标本,并保存在无水乙醇中。从所获样品中随机选取5瓶样品,手动分拣其中昆虫个体。鉴于DNA提取、易操作性等因素,本研究仅选取个头较大的个体(体长>0.5 cm),以建立方法流程。随后,按形态特征将所选样品初步分成不同的类群(即非严格的“形态种”),并对每个类群选取1~3头用无水乙醇冲洗,再分别取腿置于离心管中保存于-20℃下用于DNA提取。
1.2 DNA提取和PCR扩增
首先将样品取出并在室温下晾干残余酒精,再用液氮速冻后研磨。使用天根生化科技(北京)有限公司或凯杰(QIAGEN GmbH, Hilden, Germany)公司提供的血液和组织DNA提取试剂盒提取全基因组DNA,具体实验步骤参照相应试剂盒说明书操作。所提取的基因组DNA于-20℃保存。
本研究所有聚合酶链式反应(PCR)均使用LongGene PCR仪进行扩增。PCR总体系为30 μL:15 μL Premix PrimeSTAR HS (TaKaRa),正反引物(10 μM)各1 μL,ddH2O 10 μL ,DNA模板3 μL。针对扩增目标,即线粒体COI基因5′端长度为658 bp区域,首选通用引物LCO1490和HCO2198(Folmeretal., 1994),其具体反应程序见表1。对于以上程序扩增不成功的,则采用引物LCO1490和HCOout(Carpenter and Wheeler, 1999),并结合PCR反应程序Ⅵ(PCR Ⅵ,表1)(Huangfuetal., 2022)进行扩增。
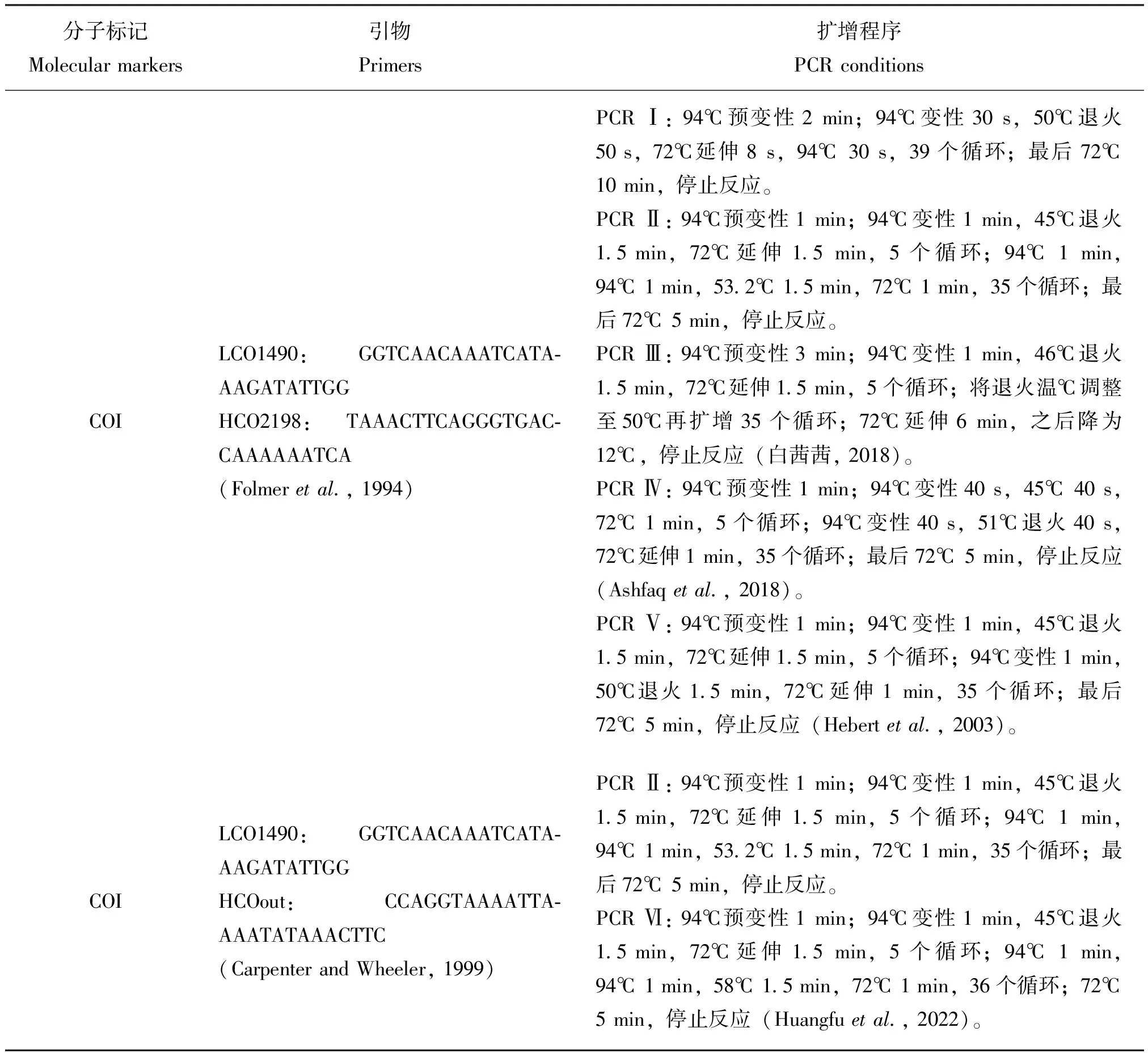
表1 PCR使用引物及反应程序
取6 μL PCR产物用于1%琼脂糖凝胶电泳,观察电泳目标条带是否明亮,并拍照记录电泳结果。将凝胶电泳检测出目标条带的样品送北京天一辉远生物科技有限公司进行Sanger测序。
1.3 数据分析
1.3.1序列处理及初步鉴定
记录测序公司返回的序列测序情况,并采用BioEdit(Hall, 1999; 2011)、MAFFT v7(https://mafft.cbrc.jp/alignment/server/)和MEGA v7 (Kumaretal., 2016)软件对序列进行处理。首先,将所有COI序列采用MAFFT进行比对,用MEGA翻译为氨基酸序列以核查阅读框是否中断,用BioEdit软件对序列进行校对、剪切,并统计序列的碱基位点情况;将序列导入到BOLD system v4在线系统,使用IDENTIFICATION程序查看物种鉴定结果;若在BOLD中无法获得分类信息,则采用NCBI中的Blast程序再次比对查询;将从BOLD和NCBI中获得的物种信息进行整合,并结合挑选样本时的形态分类情况以确定获得的序列即为目的基因片段。
1.3.2MOTU界定
上述数据库鉴定可将所有序列清晰确定到目水平(与形态分类一致),因此后续分子分类将针对涉及的昆虫各目分别展开。先将修剪好的序列采用Mothur (Schlossetal., 2009)处理成单倍型,随后采用jMOTU (Jonesetal., 2011)、ABGD (Puillandreetal., 2012)、bPTP (Zhangetal., 2013)、GMYC (Fujisawa and Barraclough, 2013) 4种方法对6个目昆虫序列分别进行物种界定,划分出MOTU。
(1)jMOTU 分析。分歧阈值设置为1~20 bp,聚集参数为97%。
(2)ABGD分析。种内差异先验值P(prior intraspecific divergence)最小值为0.001,最大值为0.1,最小相对gap宽度值X(minimum relative gap width)=0.5~1,Steps=5,Nb bins=20,基于K2P模型(Kimura, 1980)计算遗传距离。
(3)bPTP分析。首先,采用IQ-TREE(Nguyenetal., 2015)以最大似然法及默认模型构建有外群的ML系统发生树;随后,基于ML树并设置默认参数(MCMC代数为100 000,Burn-in值为0.1,seed值为123)进行物种界定结果分析。
(4)GMYC分析。基于以上构建的ML树采用r8s软件构建超度量树(ultrametric tree),继而通过R软件“SPLITS”包(Monaghanetal., 2009)中单阈值(single-threshold)GMYC模型进行物种界定分析。
基于以上方法分别获得MOTU后,采用R “clues”包的Hubert &Arabie调整兰德指数(Hubert and Arabie’s adjusted Rand index)(Millgan and Cooper, 1986; Changetal., 2010)对4种方法的划分结果进行两两比较分析,评估不同界定结果的稳定性,并结合所构建的ML树情况,选择一致性最高的分类结果确定为最终的MOTU或物种界定结果用于后续分析(Wangetal., 2019; Lietal., 2022)。
1.3.3遗传距离
根据上述确定的MOTU结果,按不同昆虫目将序列导入MEGA中,进行序列碱基含量分析;并根据物种界定所得MOTU划分情况将序列分成不同的“group”。当某MOTU中包含两条或以上序列时,根据两两平均距离(p-distance模型)计算种内遗传距离。随后,分别计算6个昆虫目的种内平均遗传距离。
2 结果与分析
2.1 序列情况及物种鉴定
本研究共计扩增479个昆虫样本,并成功获得475条COI序列,扩增成功率达99.2%。经与BOLD和NCBI在线物种数据库比对,结果显示:样本序列物种目级信息与形态学初步分类结果一致,475条序列分属于6个昆虫目(鳞翅目、双翅目、膜翅目、鞘翅目、半翅目、直翅目);其中双翅目最多,152条(占32.4%);其次为膜翅目和鳞翅目,均超过100条;鞘翅目、半翅目和直翅目均不到40条。物种数据库鉴定到科水平的COI序列共计205条,其中数量占优势的双翅目鉴定到科共70条(46.1%),属于14科;其次为鳞翅目69条(58.5%),属于15科;膜翅目46条(36.5%),属于7科;最少为直翅目,仅5条,属2科(表2)。
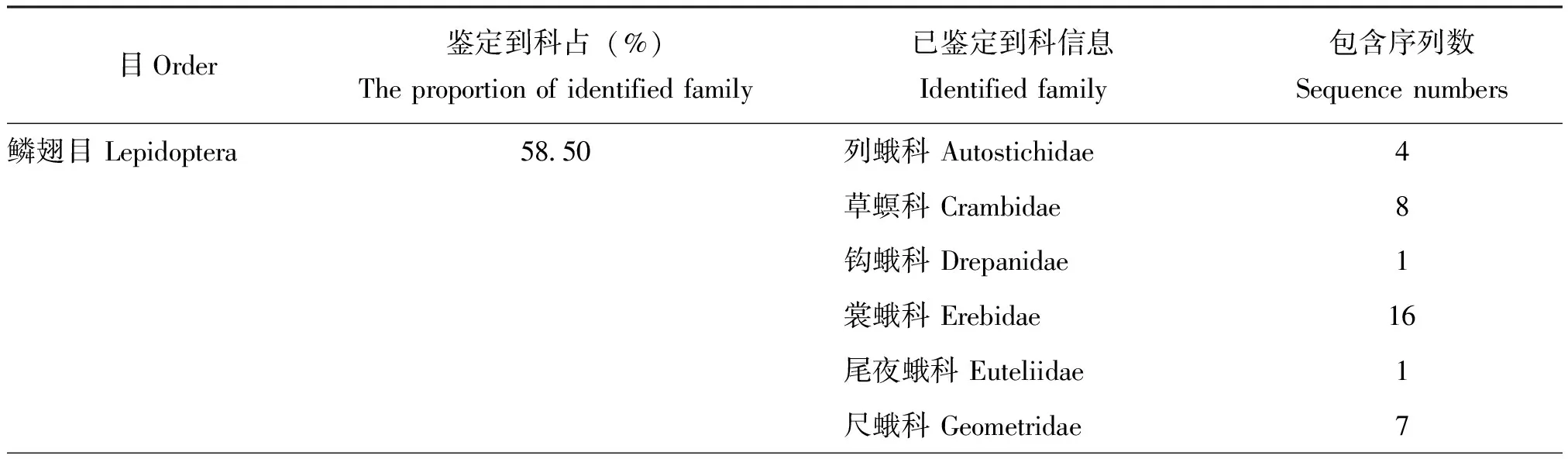
表2 不同目昆虫物种鉴定
碱基组成计算结果显示:6个目昆虫的序列碱基含量均具有明显的AT偏向性。其中,半翅目、鳞翅目、膜翅目和双翅目等4个目的A+T含量均比G+C含量高出2倍多,膜翅目A+T含量最高,达72.6%,直翅目最少为64.3%(表3)。
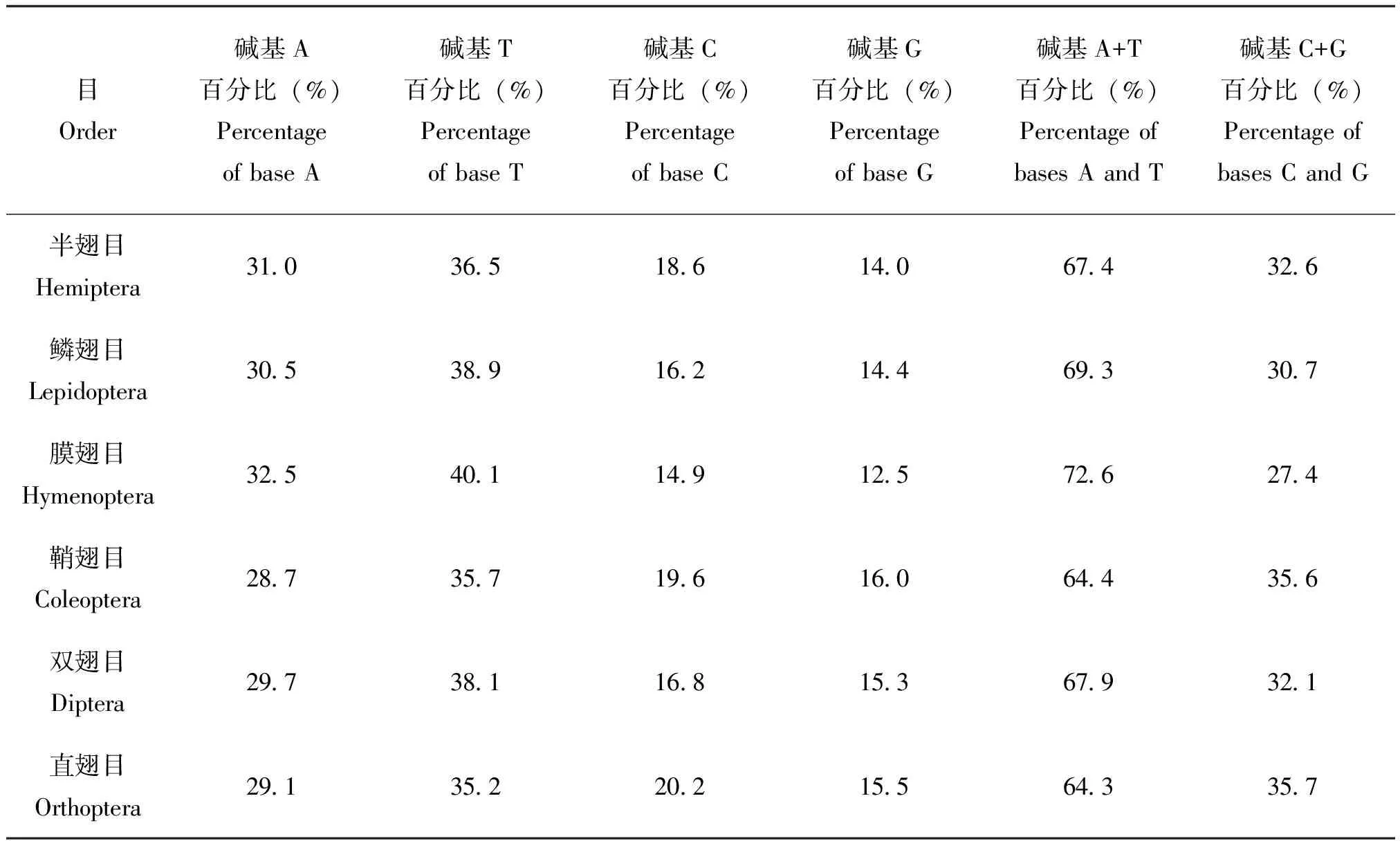
表3 不同目昆虫COI序列碱基组成信息
2.2 物种界定
所获序列经比对修剪后433条序列(占90.4%)用于物种界定分析。各昆虫目序列数从高到低依次为双翅目、鳞翅目、膜翅目、鞘翅目、半翅目和直翅目。采用jMOTU、ABGD、bPTP和GMYC这4种方法进行界定共得到288个MOTU,其中鳞翅目获得的MOTU最多,半翅目、鞘翅目和膜翅目获得MOTU均超过样本量的60%,双翅目和直翅目的MOTU不到样本量的50%(表4)。
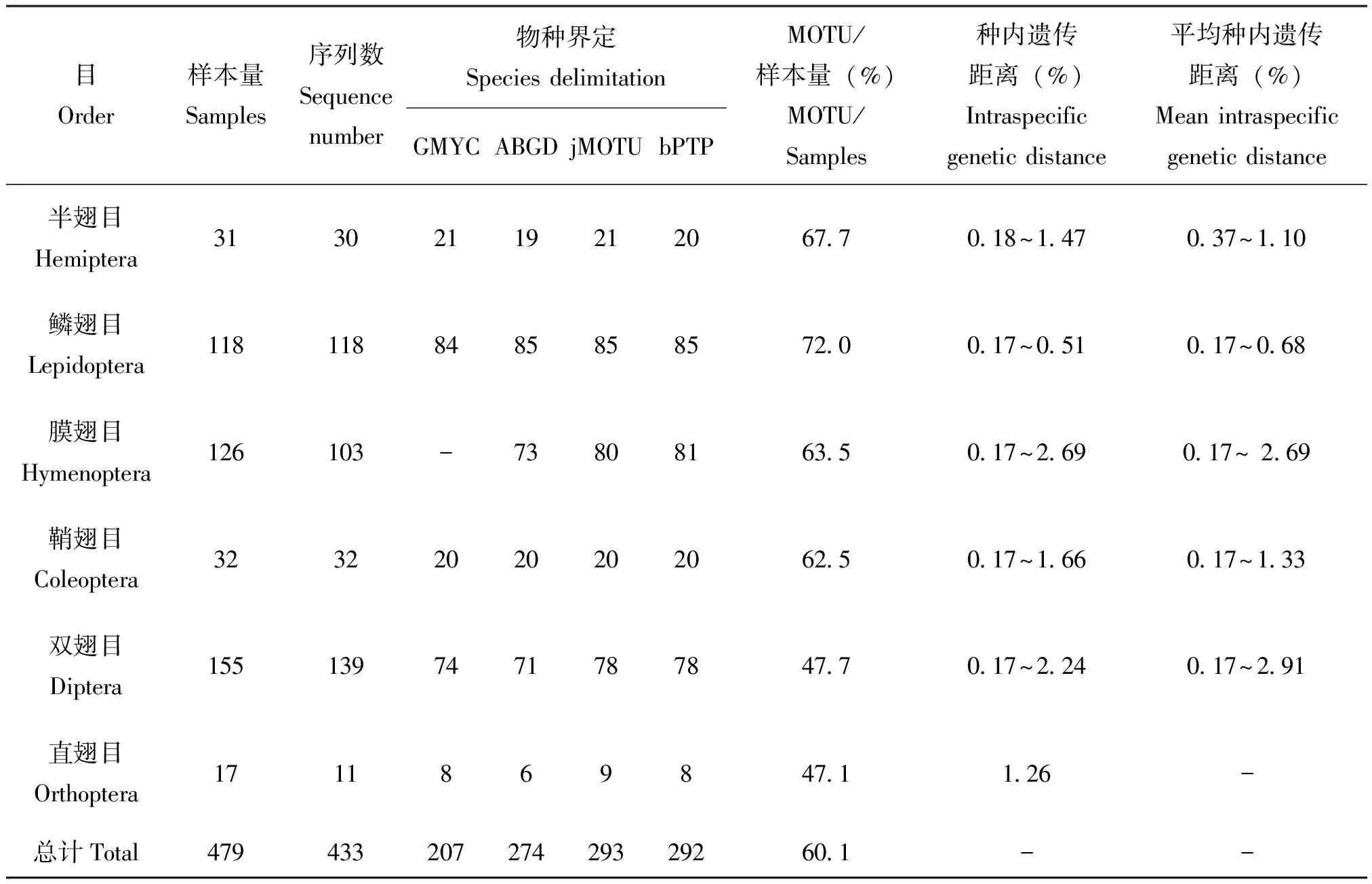
表4 各目昆虫物种界定及种内遗传距离统计情况
具体而言,鞘翅目昆虫用4种方法界定的结果完全一致。鳞翅目昆虫除用GMYC方法界定结果少一个MOTU外,其余3种方法界定结果完全一致。半翅目昆虫的jMOTU和GMYC方法界定结果完全一致,而ABGD和bPTP方法存在两处差异,涉及5条序列。bPTP和GMYC对直翅目昆虫的界定结果完全一致,但ABGD和jMOTU方法存在两处差异,涉及5条序列。4种方法对双翅目昆虫的结果均不一致,存在6处差异,涉及28条序列。GMYC外的另外3种界定方法对膜翅目昆虫界定结果均不一致,存在7处差异,涉及16条序列(图1)。根据R “clues”包Hubert &Arabie调整兰德指数对不同方法界定结果的稳定性评估情况,选取一致性最高的为最终结果。因此,鞘翅目昆虫基于4种方法划分为20个MOTUs;鳞翅目昆虫基于ABGD、jMOTU、bPTP方法划分为85个MOTUs;半翅目昆虫基于jMOTU和GMYC方法划分为21个MOTUs;直翅目昆虫基于bPTP和GMYC方法划分为8个MOTUs;双翅目昆虫基于GMYC的界定结果,划分为74个MOTUs;膜翅目昆虫基于jMOTU方法的界定结果划分为80个MOTUs。

图1 不同物种界定方法对不同目昆虫的物种界定结果Fig.1 Species delimitation results from different methods for different insect orders注:图中仅显示4种方法划分MOTU有差异的部分序列情况。Note:Figure only shows partial sequences for which species delimitations from the four methods differed.
2.3 遗传距离
基于物种界定所得MOTU计算种内遗传距离显示,不同昆虫类群的种内遗传距离最小值差异很小,鳞翅目、膜翅目、鞘翅目、双翅目均为0.17%;但最大值差异较大,从0.51%至2.69%(图2,表4)。不同昆虫类群的种内遗传距离中,膜翅目种内遗传距离均值及标准偏差最大(0.89%±0.87%,mean±SD),其次为双翅目(0.73%±0.58%)、半翅目(0.71%±0.31%)和鞘翅目(0.60%±0.47%),鳞翅目的最小(0.28%±0.20%)(图2)。
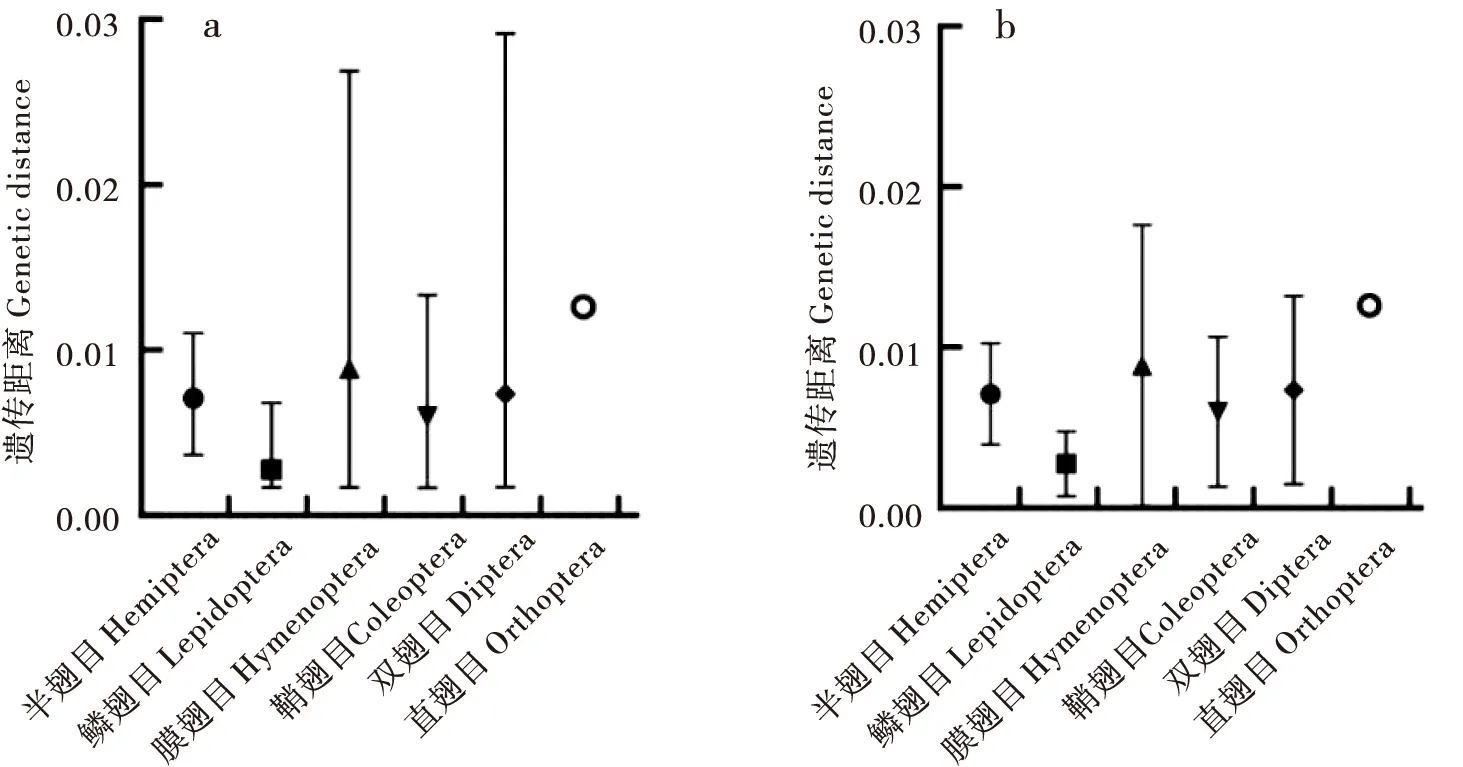
图2 不同目昆虫种内遗传距离的平均值、最大值和最小值(a)、平均值±标准差(b)Fig.2 Average, maximum and minimum value (a) and mean±SD (b) of the intraspecific genetic distance of different insect orders
3 结论与讨论
DNA条形码通常被认为是一种经济、简便、可靠的分子鉴定工具,在后生动物类群中具有广泛的适用性(Hebertetal., 2004a; Hebert and Gregory, 2005; Virgilioetal., 2010)。本研究对测得的COI条形码序列与在线物种库进行了比对鉴定,并基于划分的MOTU进行种内遗传距离分析。结果表明:不同的昆虫类群的种内遗传距离,虽然整体在一定范围内(如小于3%),但呈现一定程度的差异。因此,虽然曾有研究提出10倍法则或3%的遗传距离阈值作为物种水平差异诊断的标准(Hebertetal., 2003; Hebertetal., 2004b; Waugh, 2007),但是昆虫分子分类不能简单地依赖于某具体距离阈值。本研究计算了6个昆虫目的种内遗传距离,发现种内遗传距离最小值小于1%,而且鳞翅目、双翅目、膜翅目、鞘翅目昆虫的种内遗传距离范围具有相似性;双翅目、膜翅目昆虫的种内遗传距离和种内平均遗传距离最大值范围均大于2%。这些均与Hebert等(2003)所认为的种内遗传距离多小于1%的说法不完全一致。此外,Zhang &Bu (2022)对BOLD数据库中64 414种昆虫进行遗传距离分析发现:大约四分之一的昆虫物种存在较高的遗传变异(>3%)。然而,新近的研究中,Sharkey等(2021)依然使用2%的遗传距离阈值开展膜翅目昆虫物种划分。显然,在对昆虫进行分子分类研究中针对不同昆虫类群的划分方案还存在争议。本研究也再次证明不同的昆虫类群遗传距离存在差异性,使用特定遗传距离阈值进行物种划分时需要慎重考虑。
其实,DNA条形码用于昆虫物种鉴定在实验环节还受到取样、DNA提取、PCR扩增和测序等因素的影响。其中,非常关键的环节是使用引物扩增目的基因片段。本研究以昆虫线粒体COI条形码扩增的常用引物LCO1490+HCO2198为主,以LCO1490+HCOout作为补充进行扩增,总体获得了较高的成功率,达99.2%。不同目昆虫表现出一定差异:鳞翅目、鞘翅目、双翅目扩增成功率都达100%,而半翅目的扩增成功率只有96.8%。值得一提的是,对于双翅目昆虫使用引物LCO1490+HCOout将扩增成功率从61%提高到了98%。
同时,尽管BOLD和NCBI数据库都包含有大量的COI数据,但本研究获得的6个昆虫目400余条COI序列与在线物种数据库进行比对鉴定时,能注释到科及属、种水平的尚不足50%。Virgilio等(2010)对DNA条形码在鳞翅目、双翅目、膜翅目、鞘翅目、直翅目、半翅目这6个目昆虫中的性能比较研究中,也证实了在缺少数据库资料作参考的情况下,会影响DNA条形码的物种鉴定效率。Chesters &Zhu (2014)发现现有的物种数据库仍然有较大的物种信息缺口,需要不断完善。因此,昆虫多样性研究需要分类学者提供足够的物种信息,才能在测序技术和分析效率等方面有大幅提升的同时,为更加高效地利用分子物种鉴定方法(如:DNA宏条形码)提供基础。
本研究对亚热带森林中数量居多的鳞翅目、双翅目、膜翅目、鞘翅目、半翅目、直翅目这 6大目昆虫进行了DNA条形码研究,在目内科、属、种等多个水平探讨了DNA对不同昆虫类群的检测和界定效率,可以丰富本地昆虫分子数据库,为森林虫害防治提供基础数据;同时也为进一步探索基于DNA条形码的分子分类学研究提供参考。但本研究仍然存在一定的采样局限性(有的目样本只有30余个),不同物种的个体数量也分布不均衡,然而不影响DNA条形码在昆虫多样性研究工作中的物种鉴定有效性。后续的研究有必要扩大采样量,并进行更深入的探讨;同时应当在形态分类学的基础上加大对DNA条形码的研究,不断丰富DNA条形码数据库,从而可以为利用更加高效的分子分类方法开展多样性调查提供基础。
