注射用重组人生长激素治疗特发性矮小症Ⅲ期临床试验研究
2023-10-18卫海燕杜红伟利王美娜张惠文顾学范
梁 欢 盛 海 卫海燕 杨 玉 杜红伟 刘 芳 杨 利王美娜 王 莉 马 青 张惠文 顾学范
1.上海交通大学医学院附属新华医院 上海市儿科医学研究所儿内分泌遗传科(上海 200092);2.安徽安科生物工程(集团)股份有限公司 基因工程制药安徽省重点实验室(安徽合肥 230088);3.郑州大学附属儿童医院 河南省儿童医院郑州儿童医院 河南省儿童遗传代谢性疾病重点实验室(河南郑州 450000);4.江西省儿童医院内分泌遗传代谢科(江西南昌 330006);5.吉林大学第一医院小儿内分泌科(吉林长春 130021)
身材矮小是指在相似环境下,儿童身高处于同种族、同年龄、同性别健康儿童生长曲线-2 个标准差(SD)以下,而特发性矮小症(idiopathic short stature,ISS)是儿童矮小最常见的类型,亦称非生长激素缺乏性矮小,是指目前在常规检查下尚未发现可认知原因的身材矮小[1-2],约占所有矮身材儿童(身高低于正常均值2 个SD 或以上)的60.0%~80.0%[2-4],包括体质性生长及青春期发育延迟和家族性身材矮小。有资料显示ISS 发病率约为23/1000[1],累积矮小儿童数量相当多。
重组人生长激素(recombinant human growth hormone,rhGH)是促进ISS患儿增高的主要治疗手段。国外大量研究表明,rhGH 可增加ISS 患儿的年生长速率(height velocity,HV)和预测成年身高。多项临床研究证实其对ISS 最终成年身高的改善有一定疗效[2,5-7],并且其安全性与其他生长激素获批适应症相似[2]。国内亦有类似报道[8-9],但大多为回顾性和观察性研究。
基于多项大型临床试验和各种研究报道的meta分析,2003年8月美国食品药品管理局(FDA)正式批准了ISS为rhGH的治疗适应症[10],同时指出其安全性等问题与用于其他适应症并无差别。rhGH对中国儿童ISS 患者的疗效及安全性如何,尚缺乏相关的前瞻性临床研究。本研究进行了一项Ⅲ期临床试验,以评估rhGH在中国青春期前ISS患者中的安全性和有效性。
1 对象与方法
1.1 研究对象
本研究为2016年12月27日至2020年7月30日,从上海交通大学医学院附属新华医院、郑州大学附属儿童医院、江西省儿童医院、吉林大学第一医院招募青春期前ISS患儿开展的一项随机、开放、空白对照、多中心的为期52 周的Ⅲ期临床试验。研究对象纳入标准:①青春发育期前儿童(Tanner Ⅰ期),年龄>3周岁;②入组时绝对身高低于同年龄、同性别正常儿童身高均值2个SD,体态匀称;③HV≤5 cm/a;④无全身性、内分泌、营养性及染色体异常等情况;⑤1年内两项不同药物GH激发试验证实患儿血浆GH 峰值≥10 ng/mL;⑥骨龄(bone age,BA)半年内检查正常或延迟,其中女孩≤9岁,男孩≤10岁;⑦受试者及其监护人均自愿参加研究并签署书面知情同意书。排除标准:①肝肾功能异常者;②乙型肝炎病毒检测阳性者;③高度过敏体质;④患有系统性疾病或免疫功能低下者;⑤已确诊的肿瘤患者;⑥精神病患者;⑦其他类型的生长发育异常患者;⑧曾接受过可能干扰GH分泌或具有GH作用的药物治疗,或3个月内使用过其他激素治疗;⑨研究者认为不适合入选本临床试验的其他情况。
本研究遵从研究方案,赫尔辛基宣言阐明的伦理学原则和药物临床试验质量管理规范,获得医院医学伦理委员会批准(XHEC-A-2016-005,2016年)临审第(160914-190)号(PJ2016007,2017-01-004-F02),临床试验注册机构为国家药品监督管理局,临床试验登记号为CTR20130283。在入组前获得了受试者、父母或监护人的书面知情同意书。
1.2 方法
1.2.1 分组方法 根据美国FDA 的规定,用于治疗儿童ISS 的rhGH 最大剂量为0.20 IU·kg-1·d-1。中国儿科内分泌与代谢学会建议ISS 儿童应接受rhGH的剂量为0.13~0.20 IU·kg-1·d-1[11],因此本研究设置两个试验组,分别为高剂量组(0.20 IU·kg-1·d-1,每周0.46 mg/kg)、低剂量组(0.15 IU·kg-1·d-1,每周0.35 mg/kg),另设对照组。本研究采用区组随机化方法,随机编码由统计学人员采用SAS软件完成,受试者筛选合格后,研究者登陆中央随机系统DAS for IWRS系统(北京博之音科技有限公司提供),依据分配随机号,各中心竞争入组。受试者开始治疗后,试验组(低剂量组、高剂量组)睡前皮下注射rhGH[安徽安科生物工程(集团)股份有限公司生产],对照组只随访不治疗。
1.2.2 样本预算 高剂量组、低剂量组和对照组的样本量按3:3:1比例分配。基于先前的研究结果[11-12],本试验预计治疗52周结束时主要疗效指标,即身高标准差积分变化(ΔHT SDS),低剂量组为0.5,对照组为-0.01,公共SD预设为0.7,采用双侧检验,α=0.05,β=0.2(功效为80%);低剂量试验组与对照组按3:1 比例分配病例,低剂量试验组需要62例,对照组为21例。同时,本试验预计主要疗效指标ΔHT SDS,高剂量组为0.76,对照组为-0.01,公共SD预设为0.7,采用双侧检验,α=0.05,β=0.2(功效为80%),高剂量试验组与对照组按3:1比例分配病例,高剂量试验组需要30 例,对照组为10 例。另考虑脱落因素(增加10%样本量),同时在满足随机原则的前提下,本研究计划纳入研究对象168例,其中低剂量组和高剂量组各72例,对照组24例。
1.2.3 疗效评估 主要评价指标为治疗前后实际年龄的ΔHT SDS;次要疗效指标为:①年HV(cm/a),即12×(治疗结束时身高-治疗开始时身高)/完成治疗的时间(月);②骨成熟情况为骨龄变化/实际年龄变化(ΔBA/ΔCA),为治疗结束时骨龄与治疗开始时骨龄之差除以实际年龄变化。在基线、第13 周、第26 周、第39 周、第52 周共进行5 次访视。每次随访均进行体格检查、体格发育评估,并行血常规、尿常规、血生化检查,胰岛素样生长因子1(IGF-1)、胰岛素样生长因子结合蛋白3(IGFBP-3)、糖代谢指标和甲状腺功能检测。在基线、第26 周和第52 周进行骨骼X 线检测。对照组患者在基线、第26 周、第52 周随访时采集血样用于检测IGF-1、IGFBP-3。
1.2.4 安全性评估 试验药物的安全性指标评估是指记录所有观察到的或自发报告的不良事件,包括异常的临床症状和生命体征、实验室及影像学等检查结果。评估指标包括①一般指标:体温、心率、血压、呼吸、临床症状和体征;②耐受性指标:局部皮肤注射反应、体钠潴留的症状;③实验室检查异常结果:血常规、尿常规、血生化、空腹血糖、糖化血红蛋白、空腹胰岛素变化等。不良事件定义为接受研究用产品的受试者出现的任何不良的医学事件,该事件并不一定与治疗有因果关系,可以是任何不利和非预期的体征(包括异常实验室结果)、症状或与使用试验药物有时间相关性的疾病,其严重程度参考不良事件通用术语标准第5 版进行判断[13],在本研究中定义为临床试验过程中发生需住院治疗、延长住院时间、致伤致残、影响工作能力、危及生命、导致先天畸形、死亡等事件。不良反应和严重不良反应是指临床试验中的受试者出现的与试验药物、处理有关的不良事件或严重不良事件。不良事件和不良反应的主要区别在于因果关系是否确定。
1.2.5 退出和失访处理 对于试验过程中,出现退出、失访及方案偏离的受试者,保留其病例记录表,并以其最后一次的检测结果作为最终结果,对其疗效和不良反应进行全数据集分析(FAS)。为保证主要疗效指标的准确性,不纳入符合方案集(PPS)进行分析。方案偏离类型包括:受试者实验室检查、随访超窗;实验室检查漏检;即使不符合入选标准但仍参加了研究;接受错误治疗方案或使用不正确药物剂量及其他等。
1.3 统计学分析
所有数据均采用SAS 9.4 软件进行分析。分别采用FAS 分析基线数据和次要疗效指标,采用FAS和PPS分析主要疗效指标,采用安全数据集(SS)进行安全性分析。计量资料符合正态分布的以均数±标准差表示,多组间比较采用单因素方差分析,进一步两两比较采用LSD-t检验;非正态分布的以中位数(M)(P25~P75)表示,组间比较采用秩和检验。计数资料以例数(百分比)表示,组间比较采用χ2检验或Fisher精确概率法检验。以P<0.05为差异有统计学意义。
2 结果
2.1 人口统计学和基线特征
试验共筛选205例受试者,其中37例筛选失败,包括28 例违背纳入标准或符合排除标准,9 例自愿退出。入组的168 例被随机分至对照组(n=24)、高剂量组(n=72)和低剂量组(n=72)。4 个中心的入组病例分别为3、21、47 和97 例。试验过程中,共有18 例提前退出,最常见的退出原因为不愿继续治疗(n=6)或未按要求用药(n=4),见图1。基线时各组受试者的年龄、BA、性别、身高、体重、体质指数及HT-SDS、年HV、IGF-1 差异均无统计学意义(P>0.05),见表1。
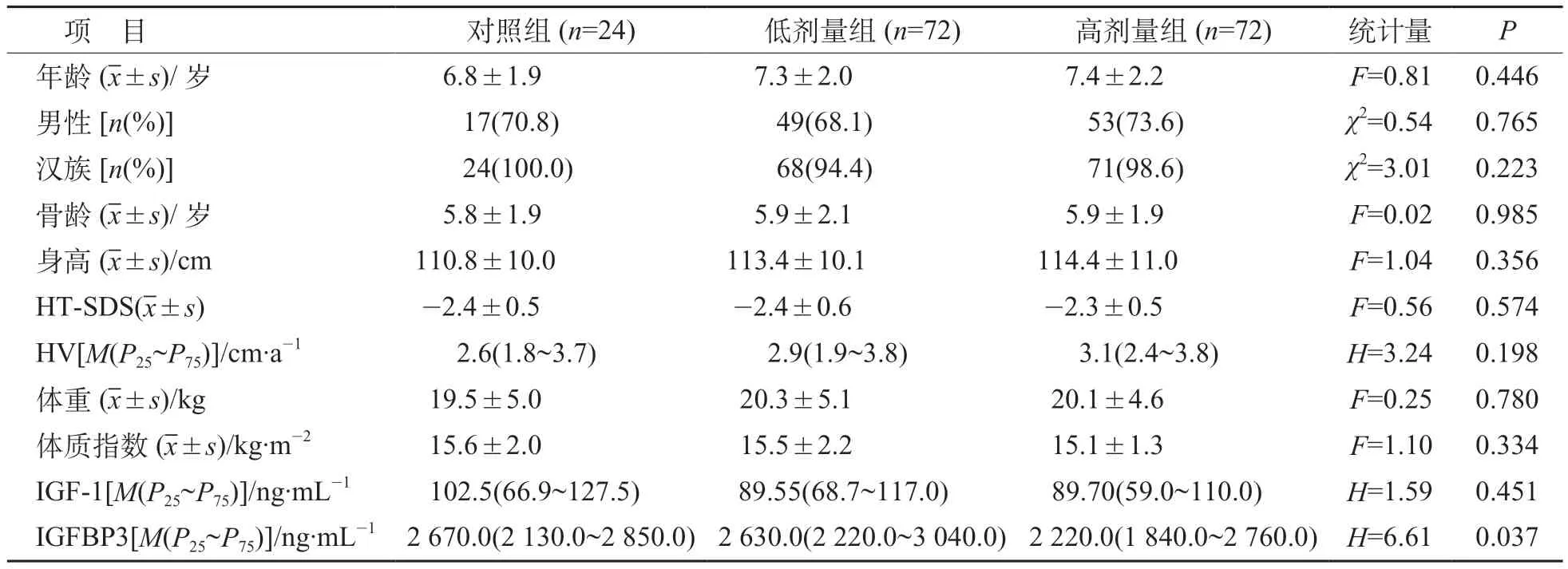
表1 人口学资料及基线特征
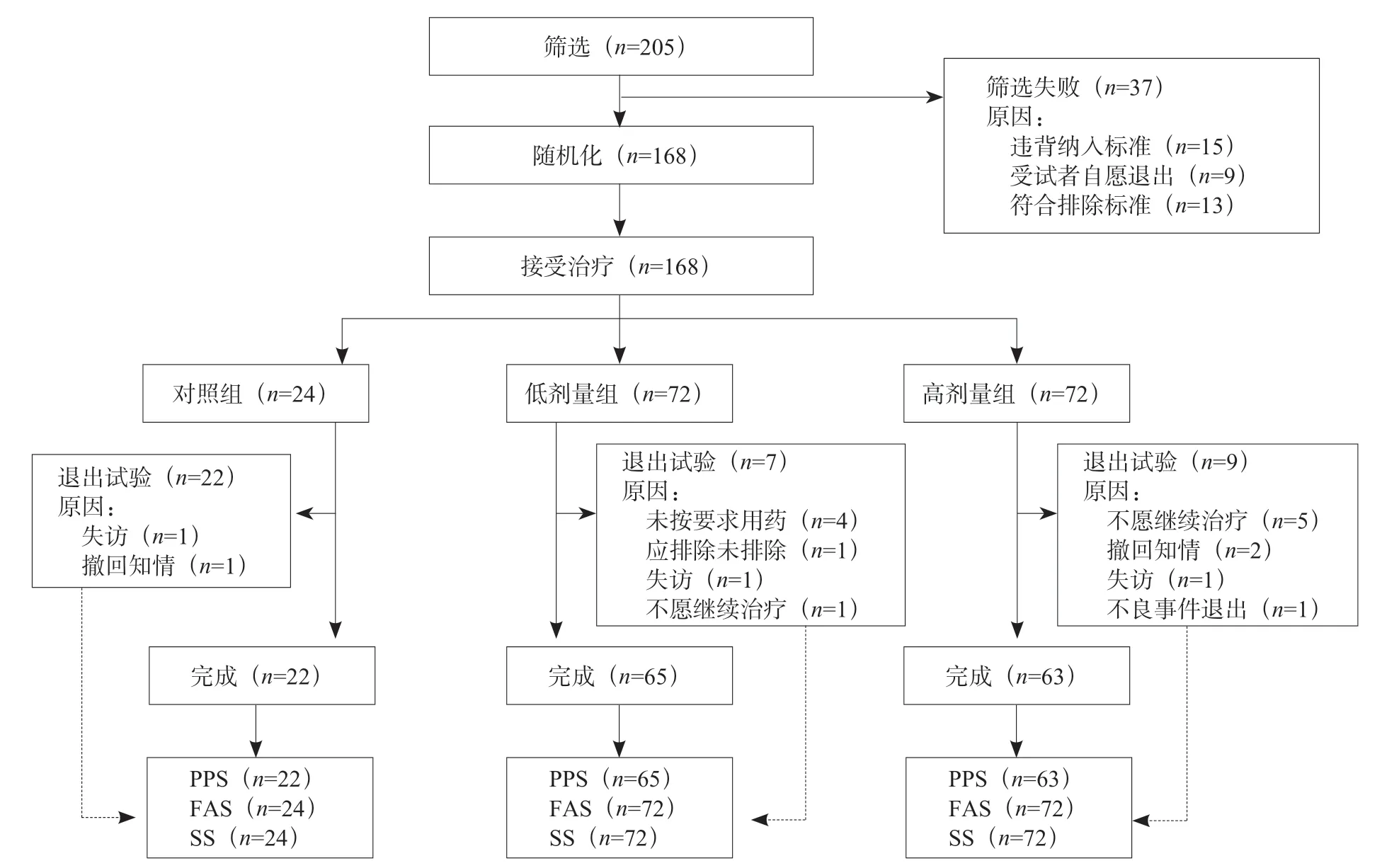
图1 试验研究对象入组情况
2.2 主要疗效指标结果
PPS 分析和FAS 分析均发现,第26、39、52 周,高剂量组、低剂量组和对照组之间ΔHT SDS的差异均有统计学意义(P<0.001);两两比较发现,高剂量和低剂量组的ΔHT SDS均高于对照组,差异有统计学意义(P<0.05),见表2。
表2 三组间疗效指标比较()

表2 三组间疗效指标比较()
注:1)与对照组比较,P<0.05
2.3 次要疗效指标结果
FAS分析发现,第13、26、39、52周,高剂量组、低剂量组和对照组之间HV 的差异均有统计学意义(P<0.001),两两比较发现,高剂量和低剂量组的HV均高于对照组,差异均有统计学意义(P<0.05)。骨成熟情况分析结果显示,第52周,各组之间ΔBA/ΔCA差异无统计学意义(P>0.05),见表2。
2.4 安全性指标
高剂量组、低剂量组和对照组之间治疗期间不良事件(TEAE)以及导致药物暂停的TEAE 发生率的差异有统计学意义(P<0.05),低剂量组与高剂量组的TEAE 发生率较高,见表3。研究过程中,各组均无死亡事件发生。
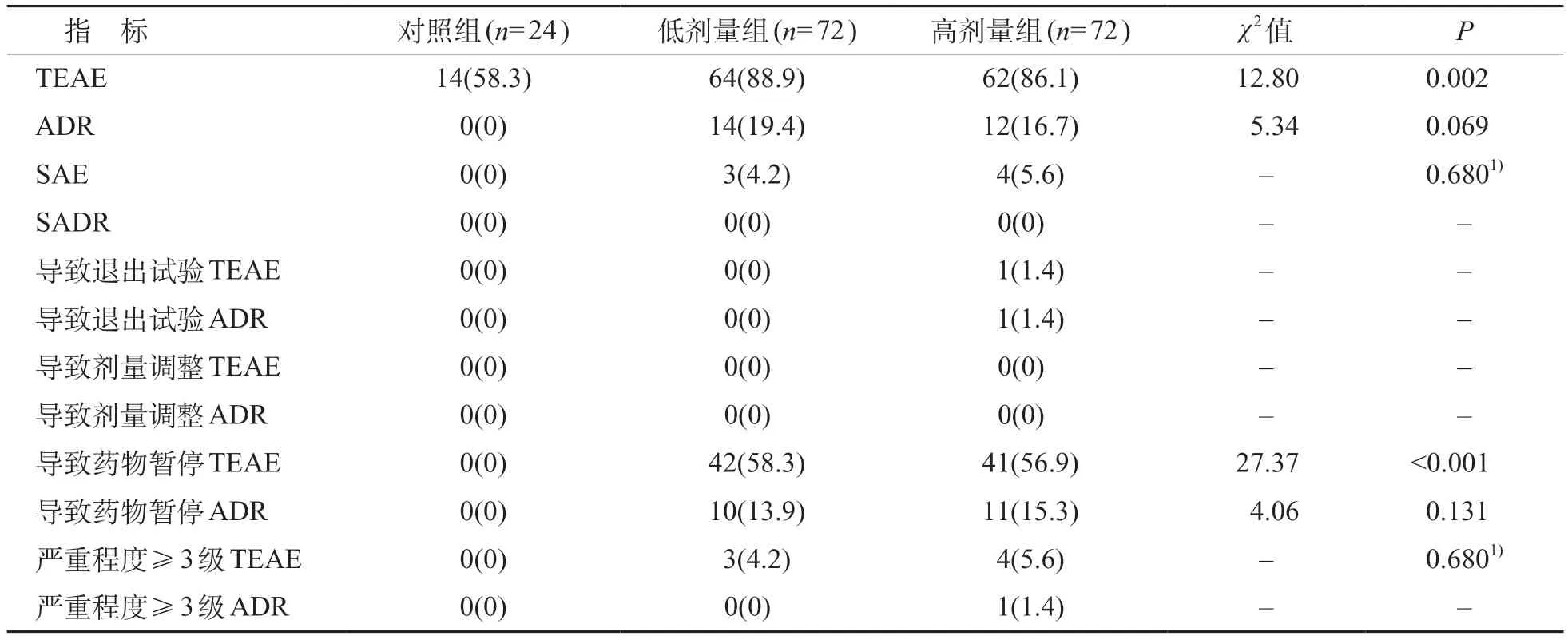
表3 研究期间不良事件分析[n(%)]
3 讨论
本试验为多中心、随机、开放、空白对照的临床研究,旨在评价注射用rhGH治疗中国青春期前ISS的有效性和安全性。研究发现,与空白对照组相比,治疗组受试者的身高增长率明显升高,提示rhGH治疗中国青春期前ISS患者52周能够明显促进其身高增长。此外,在52周时治疗组的ΔHT SDS和HV显著高于对照组,提示rhGH 用于ISS 治疗时间越长,效果可能更加明显,但长期使用的安全性和效果尚需更多的观察与评估。
rhGH 用于ISS 治疗的短期效果已有多项报道[14-17]。一项纳入了6 个随机化对照研究和4 个非随机化对照研究的meta 分析发现,rhGH 治疗ISS 患者1 年后,用药组ΔHT SDS 与对照组差值为0.6,HV差值为(2.86±0.37)cm/a[5]。近期,一项rhGH 治疗中国青春期前ISS 患者的研究也获得了类似结果,以0.05 mg·kg-1·d-1的剂量治疗52周后,用药组ΔHT SDS 为1.04±0.31,显著高于空白对照组的0.20±0.33(P<0.001);用药组的HV达到(10.18±1.47)cm/a,显著高于空白对照组[(5.81±1.68)cm/a][18]。这些研究结果表明,rhGH在ISS的短期治疗中展示出了良好的效果。此前,一项meta分析纳入了1985年至2010年的多项临床对照试验,发现rhGH治疗可以使ISS患者的成人身高增加约4 cm[19],但个体之间差异性较大,目前缺乏高质量证据和强烈推荐的循证医学标准。因此,长期使用rhGH治疗ISS对终身高的效果仍需继续观察与评估。
对骨成熟情况进行分析发现,各组受试者之间BA变化差异无统计学意义。这与此前的研究结果一致[14,20-21]。另外,我国的一项研究发现,BA/CA在基线时为0.81±0.15,治疗52 周为0.85±0.13,略有增加,但仍保持在<1的水平[17]。需注意的是,治疗第1年BA的进展与开始治疗时BA的延迟程度相关,长期治疗可能会使BA赶上CA,逐渐BA/CA 比率将增加到1[22-23]。
本研究中,以0.20 IU/kg或0.15 IU/kg的剂量每日皮下注射rhGH安全性良好,试验药与其在已获批适应症中的安全性一致,未出现新的不良反应。
综上,注射用rhGH 治疗ISS 疗效确切,患儿用药后有明显的身高增长,且未出现新的不良反应,但对终身高的获益和安全性尚需更多的观察与评估。
利益冲突声明:本文所有作者均不存在利益冲突。
