基于生物信息学和CMap数据库分析肺癌相关基因及潜在治疗药物
2023-10-13张虎黄欣李妍韩毅李羚
张虎,黄欣,李妍,韩毅,李羚
(山东第一医科大学第一附属医院<山东省千佛山医院>临床药学,山东省儿童药物临床评价与研发工程技术研究中心,山东省医药卫生临床药学重点实验室,山东 济南 250014)
肺癌是全球癌症死亡的首要原因,同时也是中国高发癌症之一[1]。近年来肺癌的治疗取得了较大的进展,比如新的治疗药物免疫检查点抑制剂的疗效已经得到了证实[2-3],但仍有患者面临着耐药、复发、转移和个体差异等问题,限制了现有药物的使用[4]。因此有效控制肺癌的发展及改善患者的总体生存期仍面临巨大的挑战,寻找新的治疗靶点和新的治疗药物也变得尤为重要。
通过整合基因相关数据库,可以更深入的研究肿瘤相关基因、分子机制,在基因层面为肿瘤的治疗寻找新的生物标志物。开发一种新的药物需要大量资金,时间和各种资源的投入,并且伴有一定的风险,而药物重定位是一种非常规的方法来识别已批准或实验药物的新适应证[5],Connectivity Map(CMap)数据库是基于基因表达谱的药物研究平台,该数据库通过大量细胞系实验将基因、药物和疾病联系起来[5]。通过CMap数据库,可筛选逆转肺癌的基因表达变化的药物。本研究通过生物信息学和CMap数据库分析,筛选出肺癌的关键致病基因和候选治疗药物,为肺癌的治疗提供了新的治疗策略。
1 材料与方法
1.1 数据的下载和DEGs的筛选从GEO(Gene Expression Omnibus,GEO)数据库(https://www.ncbi.nlm.nih.gov/geo/)中获得基因表达谱数据集,GSE89039采用GPL17077平台,其包括8个肺癌组织样本和8个正常组织样本,GSE118370采用GPL570平台,其包括6个浸润性肺腺癌组织样本和6个正常组织样本,GSE136043采用GPL13497平台,其包括5个肺癌组织样本和5个正常组织样本。采用R软件筛选DEGs,所筛选的条件为|log2FC|≥1.0,P<0.05[6],将筛选出的差异基因通过R语言(v4.2.3)可视化为火山图。
1.2 GO和KEGG的分析通过DAVID(v2021)进行GO(Gene Ontology,GO)和KEGG(Kyoto Encyclopedia of Genes and Genomes,KEGG)功能富集分析[7]。将差异基因在GO数据库进行细胞组分(cellular component,CC)、分子功能(molecular function,MF)、生物过程(biological process,BP)三方面的富集分析,在KEGG数据库进行细胞相关通路的富集分析。使用Benjamini-Hochberg方法调整P值以控制错误发现率,将调整后的P<0.05的富集设定为显著富集的临界标准[8],并且通过R语言来可视化DAVID的分析结果。
1.3 蛋白质-蛋白质相互作用分析和枢纽基因筛选将3个数据集中的交集基因放入相互作用基因/蛋白质数据库(STRING)中,高于0.4的相互作用评分设定为截止值[9]。使用Cytoscape软件(v3.9.1)构建DEG编码蛋白的网络,并将其可视化,展现重叠DEG之间潜在的相互关系[6]。
通过插件CytoHubba基于度的拓扑算法来分析网络,将具有高相互作用的前10个基因鉴定为枢纽基因。MCODE(分子复合物检测)插件来可视化和鉴定来自PPI网络的重要模块,参数设置设为默认值:degree=2,node score=0.2,k-core=2,maximum depth=100[10]。
1.4 枢纽基因的生存分析使用基因表达谱交互分析(GEPIA)中的箱形图比较每个枢纽基因在肺癌组织和正常组织之间的差异表达谱[11]。使用Kaplan-Meier分析枢纽基因上调或下调患者的总生存期(overall survival,OS)[12]。
1.5 CMap数据库筛选候选药物利用Cytoscape计算差异基因中PPI得分前100的基因集合,包括27个上调基因和73个下调基因。将这100个差异基因输入CMap(https://clue.io/)数据库,所得结果按照Score大小进行排序,筛选负相关的候选药物。
1.6 统计学分析Kaplan-Meier的生存分析,采用对数秩和检验,P<0.05具有统计学差异。在线数据库分析应用系统默认的统计学方法。
2 结果
2.1 差异表达基因的筛选在本研究中,共有38个样本,包括19个肺癌组织样本和19个正常肺部组织样本(GSE89039:8T/8N;GSE118370:6T/6N;GSE136043:5T/5N)。在基因表达测定及数据处理和标准化之后,使用Limma包对每个数据集筛选DEGs,筛选标准为:|log2FC|≥ 1.0且P值< 0.05。最终我们从GSE89039数据集中筛选了2 708个DEGs,包括1 093个上调和1 615个下调的基因(见图1A);在GSE118370中筛选了1 887个DEGs,包括814个上调和1 073个下调的基因(见图1B);在GSE136043中筛选了2 352个DEGs,包括1 164个上调和1 188个下调的基因(见图1C)。接下来,我们分别取3个基因数据集上调基因和下调基因中的重叠基因,得到150个共有上调基因和380个共有下调基因,共计530个DEGs(见图1D、E)。
2.2 差异基因的功能富集分析我们将530个DEGs进行GO和KEGG的富集分析。结果显示KEGG通路富集主要是ECM-受体相互作用,这些相互作用导致细胞活动的直接或间接控制,如黏附、迁移、分化、增殖和凋亡(见图2A)。对于GO_BP富集分析,它们在细胞黏附,细胞外基质组织,血管生成,细胞分化等富集,细胞黏附分子与肿瘤的侵袭转移密切相关,起至关重要的作用。对于GO_MF分析,它们富集在钙离子结合,细胞外基质结构成分,信号受体活性等分子功能方面。GO_CC分析显示,它们在细胞质膜的组成成分,细胞表面,转录因子复合物等部位富集(见图2B)。
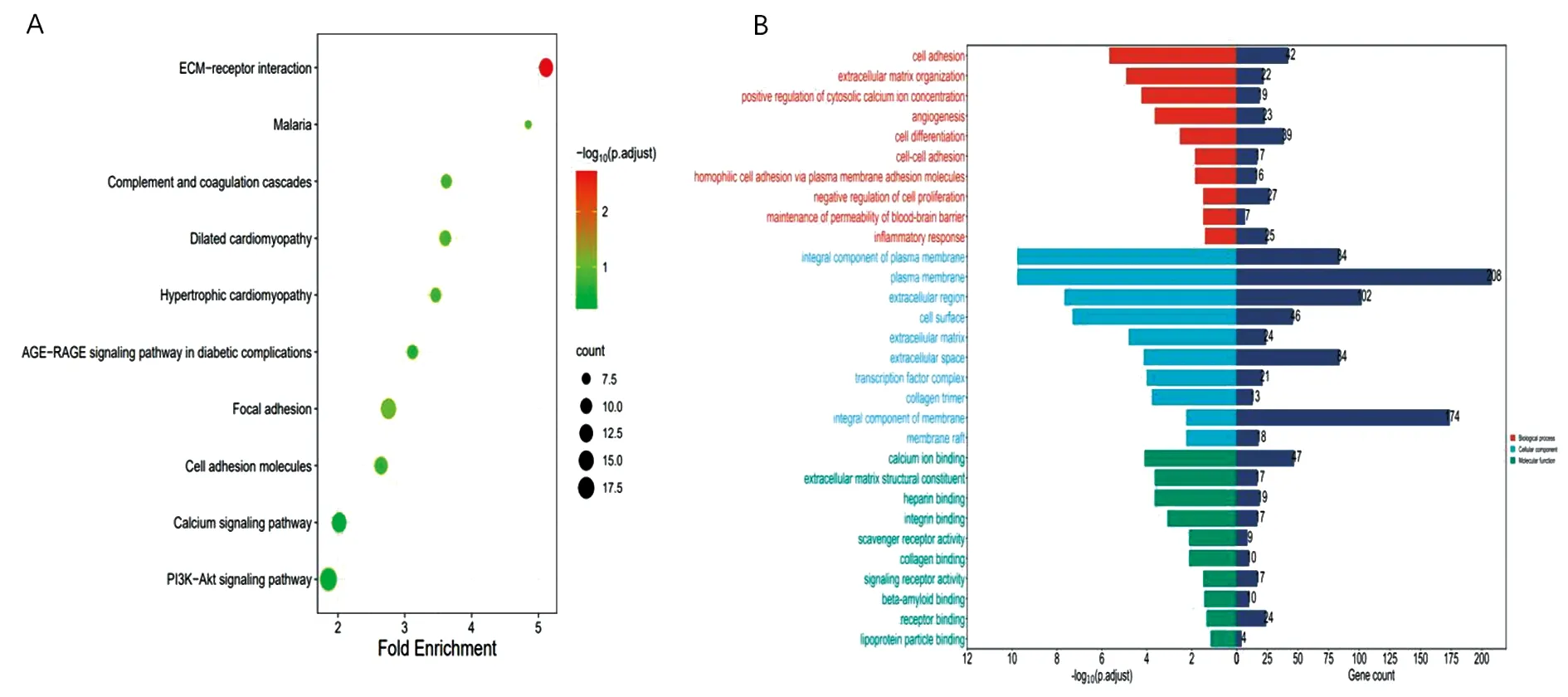
A.KEGG富集分析;B.GO富集分析,自上而下分别为生物过程(BP)、细胞成分(CC)、分于功能(MF)
2.3 PPI网络模块分析将530个DEGs构建PPI网络,由图3A可知,PPI网络由527个节点蛋白和1298个相互作用组成。通过Cytoscape的MCODE插件将整个网络聚类为12个模块,对模块得分前二进行富集分析。由图3B可知,模块1包含10个上调基因和10个下调基因,主要富集于PI3K-Akt 信号通路,该通路某些因素的突变导致的功能获得或功能缺失,引起细胞转化,同时可调节肿瘤细胞的增殖和存活,并与肿瘤的侵袭转移行为密切相关。模块2包含3个上调基因和15个下调基因,显著富集于癌症通路,RNA聚合酶Ⅱ启动子pri-miRNA转录的正向调节,转录因子复合物等,与肿瘤密切相关。
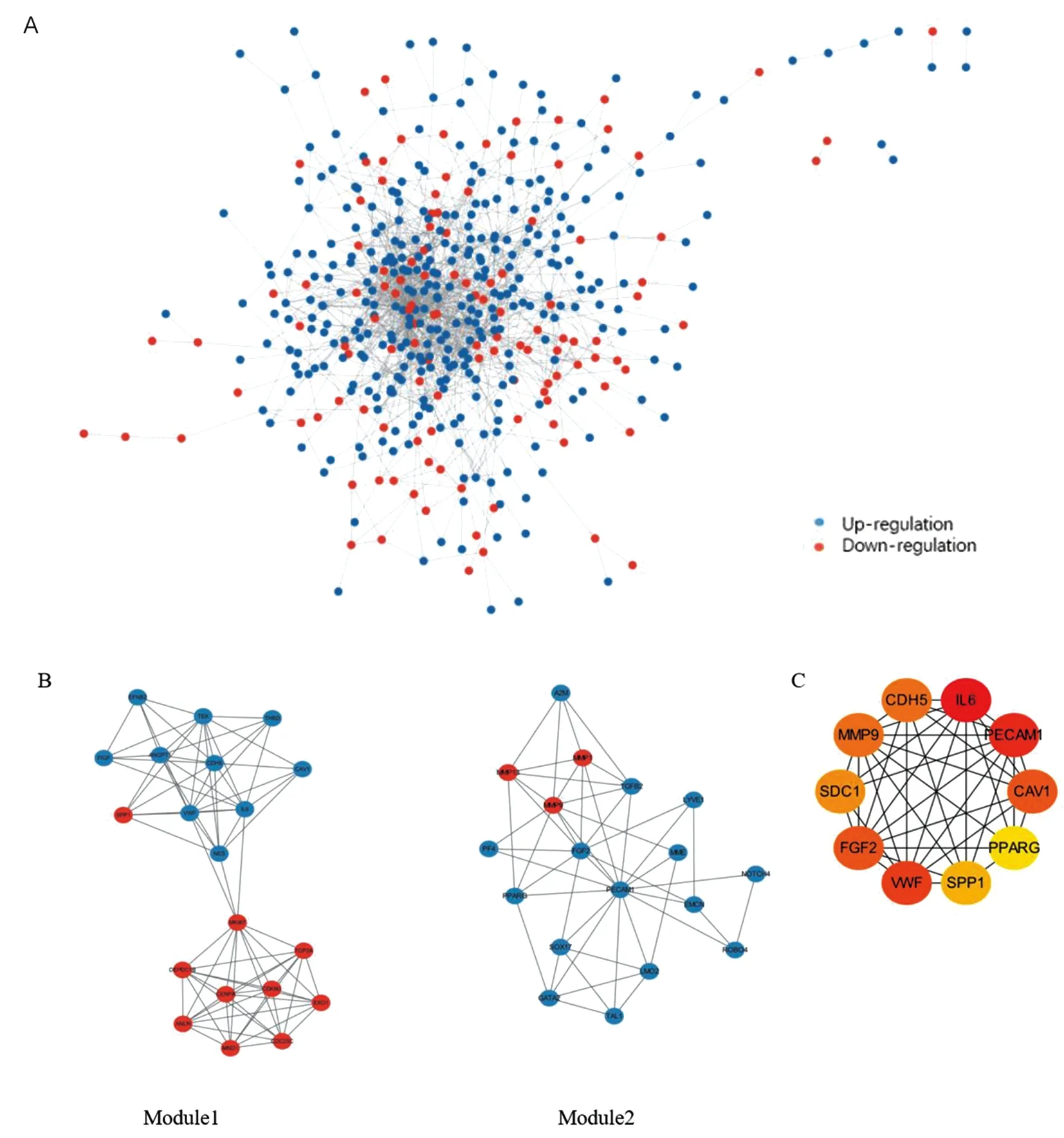
A.肺癌相关网络;红色表示常见的上调基因,蓝色表示常见的下调基因;B.差异基因中的前两个基因模块;C.肺癌相关差异基因的前10个枢纽基因模块
通过Cytoscape的CytoHubba插件在重叠DEGs中选择枢纽基因。筛选前10个基因作为枢纽基因,包括IL-6、PECAM1、VWF、FGF2、CAV1、MMP9、CDH5、SDC1、SPP1和PPARG,10个枢纽基因之间的PPI连接见图3C。
2.4 枢纽基因的鉴定接下来,我们使用GEPIA比较10个枢纽基因的差异表达(见图4)。在这10个枢纽基因中,MMP9、SDC1和SPP1在肺腺癌(LUAD)和肺鳞状细胞癌(LUSC)中均上调表达,其余7个基因在LUAD和LUSC均下调表达,与3组数据集中基因表达一致。为了检验这些基因的预后价值,我们通过GEPIA分析这些枢纽基因相关的总生存期(overall survival,OS),结果显示SPP1对OS具有显著影响(见图5)。

图5 SPP1的总生存期(OS)曲线
使用GEPIA中唯一的肿瘤数据对十个枢纽基因进行多基因比较分析。在十个基因中,SDC1和SPP1具有较高的表达水平,其次是CAV1、PECAM1、VWF、MMP9、CDH5、PPARG、IL-6、FGF2(见图6)。

图6 10个肺癌候选生物标志物的多基因比较分析
2.5 CMap数据库筛选候选药物CMap数据库包含广泛的小分子库,药物的高阳性评分表明药物可以引起或加剧疾病状态,而高阴性评分表明药物能够减轻甚至逆转该疾病状态[12]。我们将27个上调基因和73个下调基因输入CMap数据库中,筛选FDA批准上市的得分前8的药物,具体见表1。

表1 CMap筛选的抗肺癌候选药物
3 讨论
通过对肺癌细胞与正常细胞之间的基因测序,对肺癌作用机制的分析和理解更加深入,促进了分子诊断和靶向治疗研究的进展,因此我们通过生物信息学分析来寻找肺癌组织样本中有明显差异的生物标志物,为肺癌的治疗寻找新的思路与方案。在本研究中,我们建立了差异基因的PPI网络,筛选出基因之间相互作用得分前十的差异基因作为与肺癌发生发展显著关联的枢纽基因,验证枢纽基因在肺腺癌与肺鳞癌样本组织中的表达情况,发现除了SDC1在肺腺癌样本中无显著差异外,其他枢纽基因均与正常肺部组织样本基因表达量存在统计学差异。之后在基因的预后价值分析中,SPP1低表达患者的总生存期显著高于SPP1高表达患者。另外,有报道表明[13],非小细胞肺癌中循环SPP1水平或肿瘤细胞中SPP1表达水平的升高与预后不良有关。Yi等[14]研究也发现SPP1通过上调COL11A1的表达促进细胞迁移和侵袭,将SPP1作为肺腺癌转移和预后的潜在生物标志物,并且在化疗治疗的晚期非小细胞肺癌患者中,SPP1编码的骨桥蛋白在血浆中低水平表达与临床预后的改善显著相关[15],这与我们的研究一致。
药物治疗疗效的个体差异和耐药性的出现限制了现有药物的使用,且现有化疗药物不良影响较大,降低了癌症患者的生活质量,因此,这也增加了对新的候选药物的需求。然而研究和开发一种全新的药物需要大量的金钱和时间,需要广泛的进行细胞和动物研究,以及在人体进行各种安全性和有效性的临床试验[16],而药物重新定位具有成本低、风险小、周期短等优势,现已成为国内外各医药机构药物研发的重要策略[17]。在本研究中,我们将PPI中基因相互作用得分前100的基因输入到CMap数据库中,筛选出8种经FDA批准上市的药物,其中有7种为抗肿瘤药物,分别为克拉屈滨、氯法拉滨、替尼泊苷、拓扑替康、阿糖胞苷、吉西他滨和放射菌素;剩余一种非抗肿瘤药物为阿糖腺苷。拓扑替康和替尼泊苷为拓扑异构酶抑制剂,前者适用于肺癌的治疗[18],后者用于治疗恶性淋巴瘤、胶质母细胞瘤[19]。3种核糖核苷酸还原酶抑制剂分别为吉西他滨、氯法拉滨和阿糖胞苷,吉西他滨是治疗非小细胞肺癌有效的一线药物[20],氯法拉滨和阿糖胞苷共同适用于白血病的治疗[21-22]。克拉屈滨为腺苷脱氨酶抑制剂,适用于治疗多发性硬化症[23],也可以用于治疗白血病[24]。RNA聚合酶抑制剂放射菌素,抗瘤谱较窄,用于尤文氏肉瘤、睾丸癌和横纹肌肉瘤等[25],Chen等[26]研究发现放线菌素可以下调肺细胞中的Mcl-1,继而促进细胞凋亡,可能有助于新的肺癌治疗策略。
总之,通过整合GEO数据库中肺癌的DEGs,构建了一个关键枢纽基因网络来评估肺癌与正常组织样本的蛋白水平表达,并发现与肺癌相关的SPP1枢纽基因,为以后肿瘤基因水平的靶向治疗提供了新的理论依据。与此同时,我们筛选了8种具有潜在肺癌治疗作用的候选药物,为下一步的体内外实验奠定了良好的基础,也为寻找肺癌的治疗方案提供了新的思路。
