LC-MS/MS检测动物源性食品中12种激素类、硝基咪唑类和酰胺醇类药物残留
2023-10-12佟芳荻潘晓敏邱国林林佳顺
佟芳荻,潘晓敏,邱国林,林佳顺
(深圳市中鼎检测技术有限公司,广东深圳 518000)
在现代养殖业中,激素的使用可起到改善产品性状的作用,促进动物生长的同时提高生产效率[1]。而使用抗生素药物可有效降低养殖成本,使动物源性食品供应大幅增长。但激素类药物会影响人体正常代谢功能,导致早熟、发育异常等疾病[2];硝基咪唑类药物对人有致癌、致畸、致突变作用[3];酰胺醇类药物可抑制骨髓造血功能,引起粒细胞及血小板生成减少,导致不可逆的再生障碍性贫血。以上3类药物在《食品安全国家标准 食品中41种兽药最大残留限量》(GB 31650.1—2022)[4]以及中华人民共和国农业农村部第250号公告[5]中禁止使用或限制使用。针对这3类药物,我国出台了一系列检测标准,检测方法有气相色谱法[6]、酶联免疫法[7-8]、高效液相色谱法[9]和液相色谱串联质谱法[10-12]。动物源性食品基质复杂,其中激素类药物、硝基咪唑类药物和酰胺醇类药物含量极低,酶联免疫法和高效液相色谱法选择性和特异性较差,对于多残留检测有较大局限性;气相色谱法需要对目标物进行衍生,增加了检测成本;而液相色谱串联质谱法选择性和特异性好,同时灵敏度高,适合多残留检测。本研究建立了动物源性食品中激素类、硝基咪唑类和酰胺醇类药物多残留的液相色谱串联质谱检测方法,采用基质外标法定量,以提高检测效率的同时提高检测结果的准确性。
1 材料与方法
1.1 材料与试剂
牛奶、鱼肉、猪肾、鸡肉、猪肝、鸡蛋空白基质样品;地塞米松、氢化可的松、可的松、孕酮、美雄酮、睾酮、洛硝哒唑、甲硝唑、地美硝唑、氯霉素、甲砜霉素、氟苯尼考标准品,纯度大于95%,均来自德国Dr公司;乙酸乙酯(分析纯);甲醇(HPLC);乙腈(HPLC);甲酸(HPLC);一级水(GB/T 6682—2008);HLB固相萃取柱(200 mg/6 mL);0.22 μm亲水性滤膜。
1.2 仪器和设备
XEVO-TQD液相色谱串联质谱仪,Water公司;电子天平(十万分之一);电子天平(千分之一);匀浆机;涡旋混合器;冷冻离心机;氮吹仪;全自动固相萃取仪。
1.3 实验方法
1.3.1 标准溶液的配制
准确称取12种药物的标准品各10 mg,分别置于10 mL容量瓶内,用甲醇溶解并定容至刻度,配制成浓度为1 mg·mL-1的标准混合储备液。
精密量取12种药物的标准混合储备液100 μL,置于10 mL容量瓶内,用甲醇定容至刻度,配制成浓度为10 μg·mL-1的标准混合工作液。
1.3.2 样品预处理
称取5.00 g(精确至0.001 g)试样,置于50 mL离心管中,加入15 mL乙酸乙酯,均质1 min,超声提取10 min,7 000 r·min-1离心3 min,取上清液,残渣继续加入15 mL乙酸乙酯,涡旋提取10 min,7 000 r·min-1离心3 min,合并上清液。40 ℃氮吹至近干,加入3 mL 20%甲醇水溶液复溶,待净化。HLB小柱依次用3 mL甲醇和3 mL一级水活化,复溶液以1 mL·min-1的流速通过固相萃取小柱,再用3 mL 20%甲醇水溶液淋洗,真空抽干,用10 mL甲醇洗脱。洗脱液在40 ℃氮吹至近干,用50%甲醇水溶液定容至1 mL,过0.22 μm亲水性滤膜,供液相色谱-串联质谱仪测试。
1.3.3 仪器条件
(1)色谱条件。色谱柱:C18(100 mm×2.1 mm,1.8 μm);柱温:40 ℃;流速:0.3 mL·min-1;流动相A:5 mmol·L-1乙酸铵水溶液;流动相B:乙腈。梯度洗脱程序:0~0.2 min,90%A;0.2~2.5 min,90%A→2%A;2.5~3.5 min,2%A;3.5~3.6 min,2%A→90%A;3.6~7.0 min,90%A。
(2)质谱条件。离子源:ESI源;扫描方式:正/负离子模式;毛细管电压:正离子0.5 kV,负离子2.5 kV;脱溶剂温度:500 ℃;脱溶剂气流速:1 000 L·h-1;离子源温度:150 ℃;各药物的多反应监测(Multi Reaction Monitor,MRM)参数见表1。
2 结果与分析
2.1 标准曲线
精密量取一定量的地塞米松、氢化可的松、可的松、孕酮、美雄酮、睾酮、洛硝哒唑、甲硝唑、地美硝唑、氯霉素、甲砜霉素和氟苯尼考标准混合工作液,加入空白基质样品经预处理,得到标准系列混合工作液,地塞米松、氢化可的松、可的松、孕酮、美雄酮、睾酮、洛硝哒唑、甲硝唑和地美硝唑的浓度为2 ng·mL-1、5 ng·mL-1、10 ng·mL-1、20 ng·mL-1、50 ng·mL-1和100 ng·mL,氯霉素、甲砜霉素、氟苯尼考的浓度为0.2 ng·mL-1、0.5 ng·mL-1、1.0 ng·mL-1、2.0 ng·mL-1、5.0 ng·mL-1和10.0 ng·mL-1。标准系列混合工作液经液相色谱串联质谱仪测试,根据测得的峰面积(Y)与相应的浓度(X)绘制标准曲线,12种药物的相关系数在0.995~0.999,曲线方程见表2。
2.2 方法灵敏度、准确度和精密度
采用空白样品添加目标化合物的方法,经实验得到定量离子峰信噪比>10的最低浓度为方法定量限。地塞米松、氢化可的松、可的松、孕酮、美雄酮、睾酮、洛硝哒唑、甲硝唑、地美硝唑的定量限为1.0 μg·kg-1;氯霉素、甲砜霉素、氟苯尼考的定量限为0.1 μg·kg-1。空白基质添加定量限浓度目标药物的质谱图见图1、图2。
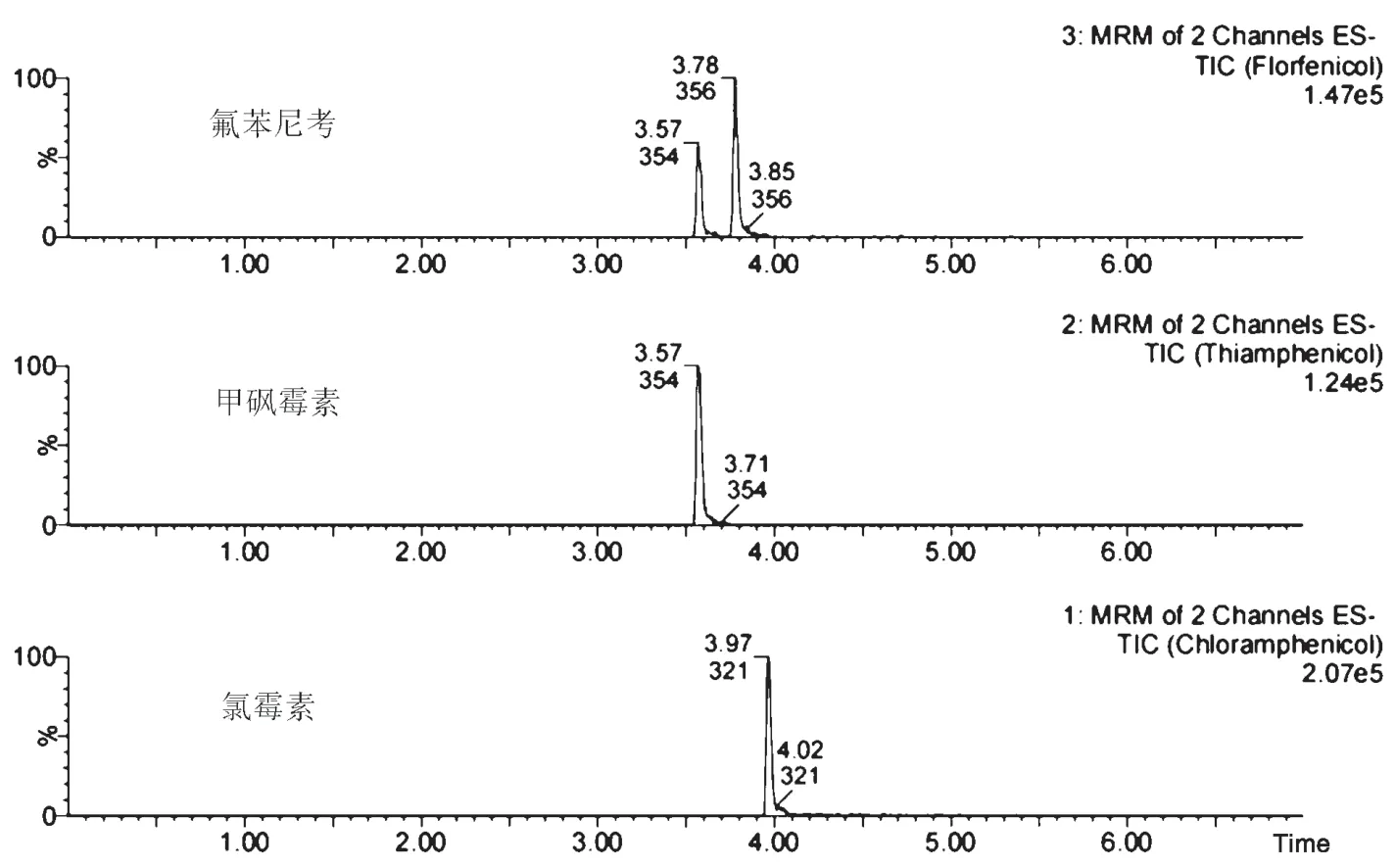
图2 氯霉素、甲砜霉素、氟苯尼考的质谱图
在鸡蛋、猪肉、猪肝、猪肾、鱼肉和牛奶6种空白基质中添加1.00 μg·kg-1、2.00 μg·kg-1、10.00 μg·kg-1的地塞米松、氢化可的松、可的松、孕酮、美雄酮、睾酮、洛硝哒唑、甲硝唑和地美硝唑,添加0.10 μg·kg-1、0.20 μg·kg-1、1.00 μg·kg-1的氯霉素、甲砜霉素、氟苯尼考,进行加标回收试验,每个浓度进行7次平行试验,结果见表3。在不同加标水平下,回收率在70%~120%,精密度<10%,回收率和精密度满足相关要求。
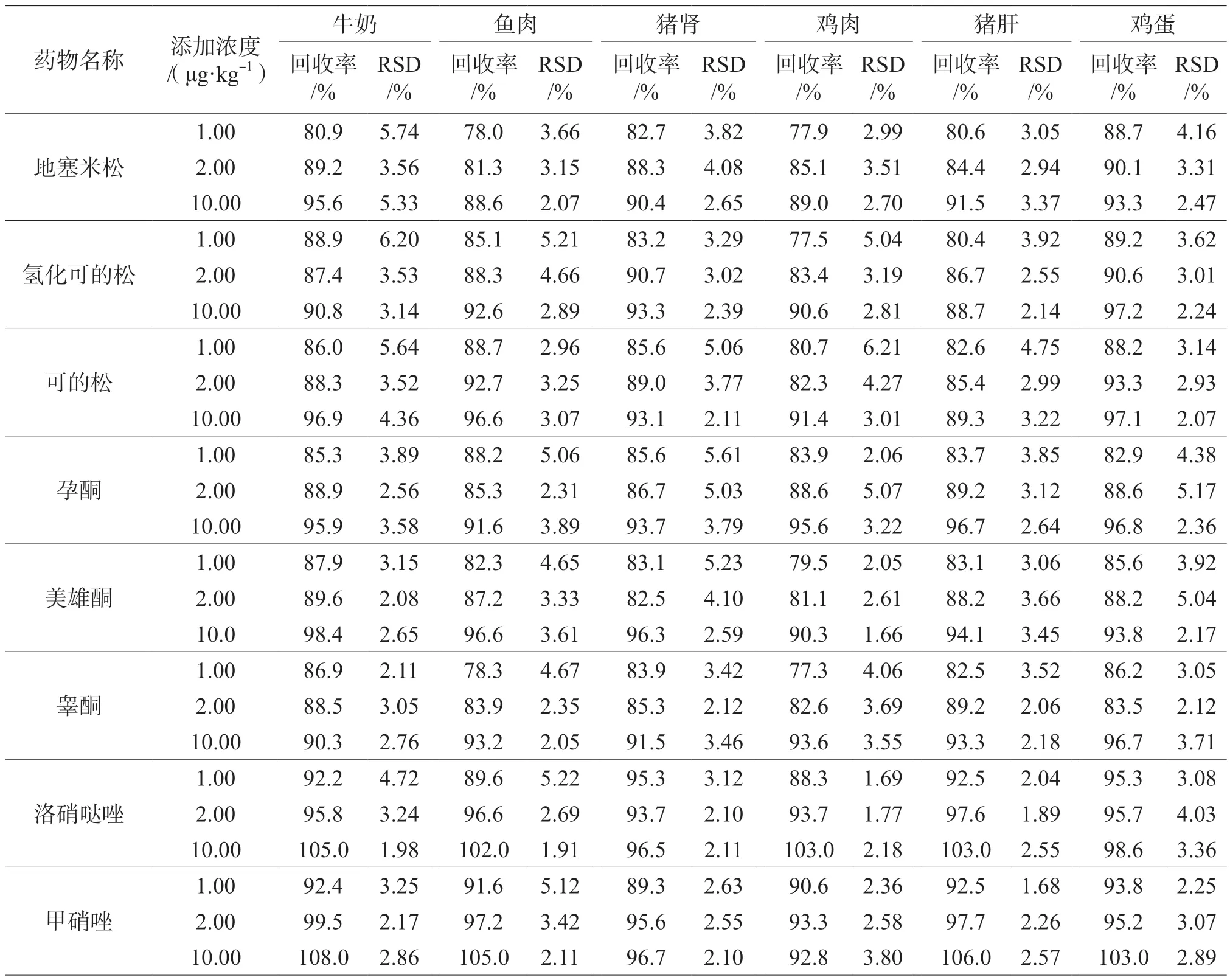
表3 加标回收率与精密度试验结果
3 讨论
(1)前处理条件的优化。本研究比较了乙酸乙酯、乙腈和甲醇3种试剂的提取效果,甲醇提取效率最低;用乙腈提取样品时,样品表面的蛋白迅速凝固,导致提取不足;乙酸乙酯提取效率最高。采用HLB固相萃取小柱进行净化,可以除去样品中的大分子蛋白杂质以及脂类,可有效减少基质效应。
(2)仪器条件的优化。本研究采用浓度为1 μg·mL-1的目标物标准工作液,分别在正负离子模式下进行全扫,优化锥孔电压,确认母离子,再对母离子进行子离子扫描,确定碰撞能等质谱参数,选择离子丰度较高的一对子离子,作为定量离子和定性离子。对流动相溶液进行优化,对比了5 mmol·L-1乙酸铵水溶液与乙腈体系和一级水与乙腈体系的目标物响应值和峰形,在流动相体系中添加少量的电解质,可提高ESI离子化效率以及减少基质效应的影响。最后选用5 mmol·L-1乙酸铵水溶液与乙腈体系对目标物进行分离。
4 结论
本研究建立了动物源性食品中激素类药物、硝基咪唑类药物和酰胺醇类药物多残留的液相色谱串联质谱检测方法。采用HLB固相萃取小柱对样品进行净化,优化流动相体系增强目标药物的响应,减少基质效应的影响,最后使用空白基质标准曲线进行外标法定量,抵消剩余基质效应的影响,使检测方法的准确度、灵敏度均能满足要求。
