HPLC-MS/MS法测定猪肉中噻虫胺残留量的不确定度评定
2023-10-12陈琳涵陈慧冰
陈琳涵,陈慧冰
(广州南沙明曦检测服务有限公司,广东广州 511400)
噻虫胺是高效、高选择性的新烟碱类杀虫剂,主要用于水稻、蔬菜、水果等作物的害虫防治[1-2]。目前,分析噻虫胺的检测方法主要有液相色谱法[3]、液相色谱-串联质谱法[4]和气质联用法[5]等,研究对象包括植物源性样品基质和动物源性样品基质。我国GB 2763—2021中规定噻虫胺在谷物和蔬菜等植物源性食品中的最大残留限量在0.01~10.00 mg·kg-1,在肉、蛋、乳等动物源性食品中的最大残留限量在0.01~0.20 mg·kg-1。目前,对猪肉中噻虫胺残留的不确定度分析鲜有报道。因此,本文采用高效液相色谱-串联质谱法测定猪肉中的噻虫胺,参照现行不确定度分析方法及要求,对其结果进行不确定度评估计算,找出影响检测结果的主要因素,为保证检测结果的准确性提供理论依据[6-8]。
1 材料与方法
1.1 材料与试剂
市售猪肉;噻虫胺标准品(农业农村部环境保护科研监测所,100 μg·mL-1,扩展不确定度为0.12 μg·mL-1);乙腈、甲醇(默克公司,色谱级);乙腈、无水硫酸镁、氯化钠(分析纯)。
1.2 仪器与设备
1290-6460液相色谱-质谱联用仪及色谱柱InfinityLab Poroshell 120 EC-C18(100 mm×2.1 mm,2.7 μm)(安捷伦公司);Centrifuge 5810 R冷冻离心机(艾本德公司);N-EVAP 32氮吹仪(Organomation公司);JJ500电子天平(双杰公司)。
1.3 实验方法
1.3.1 标准溶液的配制
(1)噻虫胺标准中间液的配制。用1 mL移液管移取1.00 mL噻虫胺标准储备液至50 mL容量瓶中,加乙腈定容至刻度,得到噻虫胺标准中间液的浓度为2 mg·L-1。
(2)噻虫胺标准系列溶液的配制。分别移取不同体积的噻虫胺标准中间液至100 mL容量瓶中,加乙腈定容至刻度,得到浓度为0.000 5 mg·L-1、0.001 0 mg·L-1、0.002 0 mg·L-1、0.005 0 mg·L-1、0.010 0 mg·L-1、0.020 0 mg·L-1、0.050 0 mg·L-1和0.100 0 mg·L-1的系列标准工作溶液。
1.3.2 样品前处理
准确称取5.00 g试样于50 mL离心管中,加入3 g无水硫酸钠,混匀,加入10 mL乙酸-乙腈溶液、10 mL正己烷,均质3 min,4 000 r·min-1离心10 min,提取液全部移至分液漏斗中,手摇1 min后静置分层,收集乙腈层,按同样操作重复提取一次。用10 mL乙腈洗涤正己烷层,手摇1 min后静置分层,收集乙腈层。合并3次收集的乙腈层,在40℃下旋蒸至近干,用乙酸-乙腈溶液定容到20 mL,取1.0 mL加适量分散固相萃取剂净化,剧烈振摇1 min,4 000 r·min-1离心10 min,取上清液用0.22 μm滤膜过滤,测定。
1.3.3 仪器条件
(1)色谱条件。色谱柱:InfinityLab Poroshell 120 EC-C18(100 mm×2.1 mm,2.7 μm);柱温:40 ℃;进样量:2 μL;流动相A:5 mmol·L-1乙酸铵水溶液(含0.1%甲酸);流动相B:甲醇;流速:0.3 mL·min-1;洗脱条件见表1。
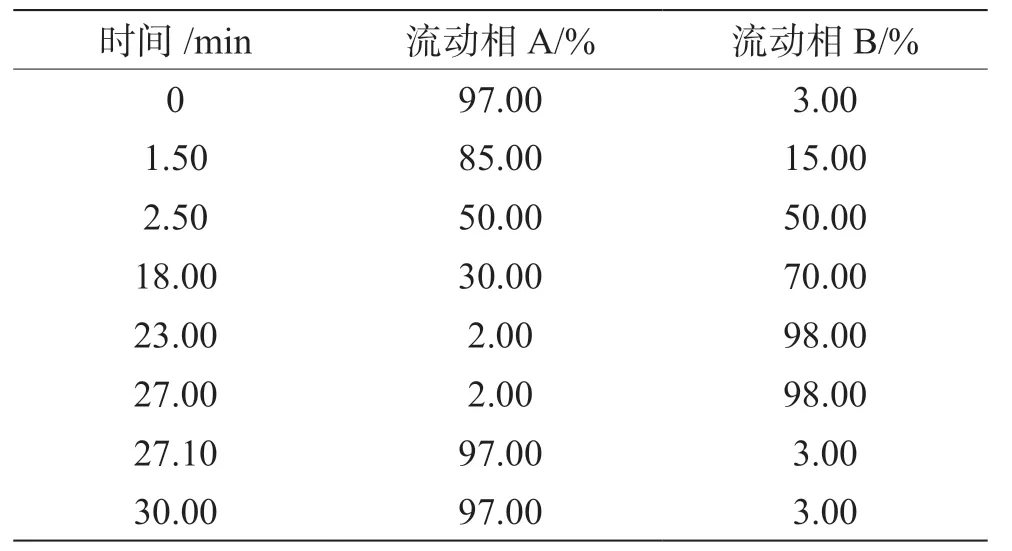
表1 洗脱条件
(2)质谱条件。电喷雾离子源:ESI+;干燥气温度:250 ℃;干燥气流量:7 L·min-1;雾化气压力:30 psi;鞘气温度:350 ℃;鞘气流量:11 L·min-1;毛细管电压:4 500 V;监测模式:多反应监测,母离子为250.2,监测离子对为169.1/132;碎裂电压:80 V;碰撞电压:10/15 V。
2 结果与分析
2.1 数学模型
噻虫胺药物含量计算公式为
式中:X为样品中噻虫胺的含量,mg·kg-1;C为样品溶液中噻虫胺的浓度,mg·L-1;V为样品定容体积,mL;m为称样质量,g。
2.2 不确定度来源分析
根据样品检测的药物含量计算公式,得到不确定度来源,见图1。
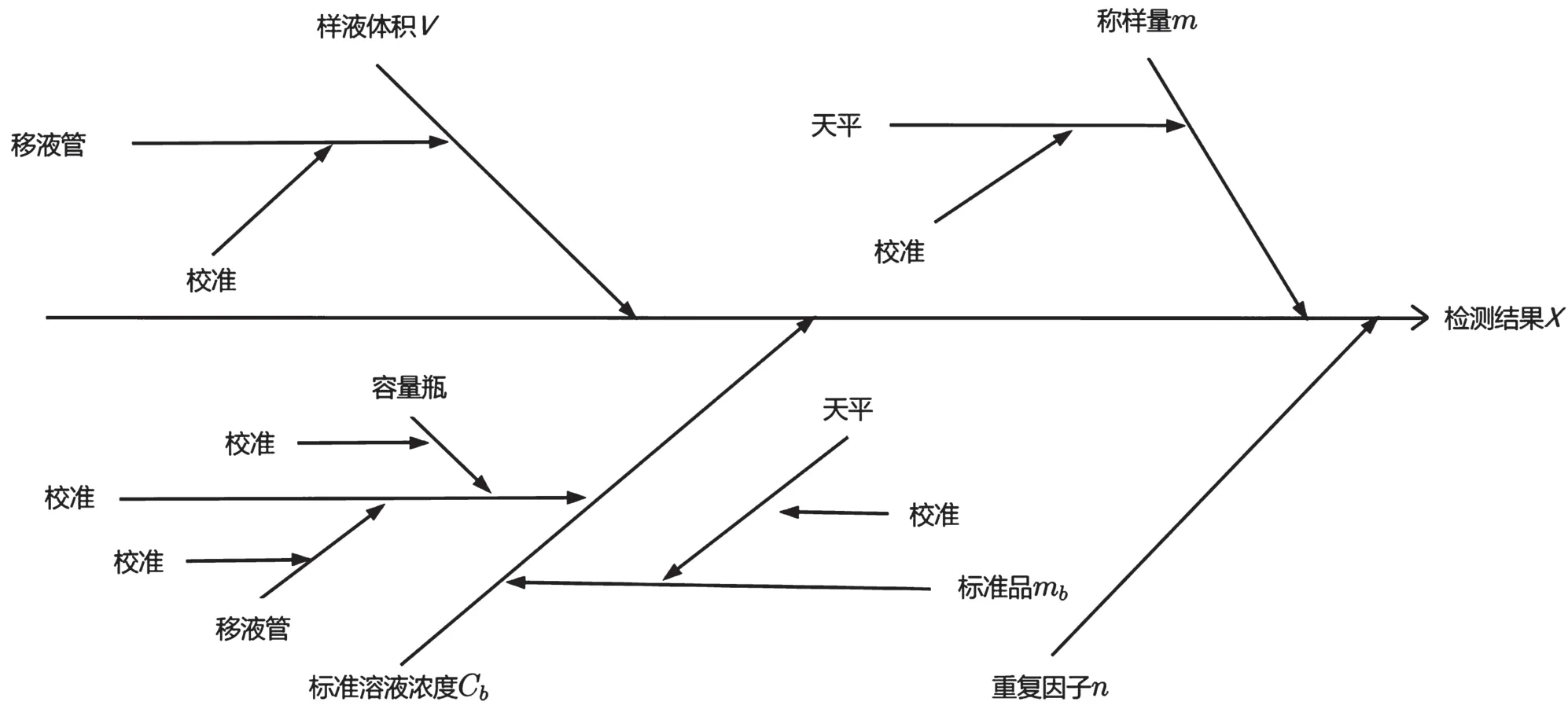
图1 猪肉样品中噻虫胺测定的不确定度来源
2.3 不确定度分量计算
2.3.1 试样溶液重复性测定引入的不确定度urel(A)
预评估时,在统计控制状态下,按照GB 23200.39—2016进行测量,对处理好的样品独立测量7次,样品含量平均值为0.003 59 mg·kg-1,单次测量标准偏差为
实际测量时将在重复性试验条件下独立测定样品2次(2个平行样)的平均值作为检测结果,平行测定的结果为0.020 44 mg·kg-1和0.019 74 mg·kg-1,平均值为0.020 1 mg·kg-1,因此测量重复性引入的标准不确定度为
测量重复性引入的相对标准不确定度为
2.3.2 称量样品质量时电子天平引入的不确定度urel(B1)
称量使用的电子分析天平(精度为0.01 g),查电子天平检定证书,U=0.02 g,k=2,因此单次称量标准不确定度为u1==0.01 g。样品称量m=5.00 g,因此相对标准不确定度为urel=×100%=0.200%。由于称量时采用差减法,一次作为空盘,另一次作为称重,每一次称重均为独立的观测结果,互不相关,由此得到称量样品质量时电子天平引入的合成相对标准不确定度为
2.3.3 标准物质浓度引入的不确定度urel(B2)
根据标准物质证书提供的信息,噻虫胺标准物质的扩展不确定度为0.12 μg·mL-1,k=2,标准值为100 μg·mL-1,则标准物质浓度引入的相对扩展不确定度为
相对标准不确定度为
2.3.4 标准溶液配制引入的不确定度urel(B3)
(1)移取1 mL噻虫胺标准储备液引入的不确定度urel(V1)。该步骤使用的是经检定合格的1 mL A类分度吸量管,经查检定证书,U=0.003 5 mL,k=2,因此单次移取溶液的标准不确定度为
移取1 mL液体引入的相对标准不确定度为
(2)噻虫胺中间液定容50 mL体积引入的不确定度urel(V2)。该步骤使用的是经检定合格的50 mL A类单标线容量瓶,经查检定证书,U=0.030 mL,k=2,因此容量瓶引入的标准不确定度为
50 mL容量瓶引入的相对标准不确定度为
(3)移取0.5 mL噻虫胺中间液引入的不确定度urel(V3)。该步骤使用的是经检定合格的0.5 mL A类分度吸量管,经查检定证书,U=0.002 5 mL,k=2,因此单次移取溶液引入的标准不确定度为
移取0.5 mL液体引入的相对标准不确度为
(4)噻虫胺上机液定容至100 mL引入的不确定度urel(V4)。该步骤使用的是经检定合格的100 mL A类单标线容量瓶,经查检定证书,U=0.050 mL,k=2,因此容量瓶引入的标准不确定度为
100 mL容量瓶引入的相对标准不确定度为
故标准溶液配制引入的相对标准不确定度为
2.3.5 试样溶液制备引入的相对不确定度urel(B4)
移取提取溶液20 mL使用的是经检定合格的20 mL A类分度吸量管,经查检定证书,U=0.050 mL,k=2,因此单次移取的标准不确定度为
移取1 mL液体引入的相对标准不确定度为
故试样溶液制备引入的相对不确定度为
2.3.6 标准物质校准曲线引入的不确定度urel(B5)
对系列标准工作溶液分别进行1次测定,峰面积响应值分别为862、1 757、3 057、6 537、14 765、28 284、68 697和143 851。以溶液浓度为横坐标(x),峰面积响应值为纵坐标(y)进行曲线拟合,得到校正曲线方程为y=1 412.895 693x+184.983 460,R2=0.999 130 34。校准曲线引入的标准不确定度为
式中:SR为残差标准差,SR=0.674 7;b为回归方程中的斜率,b=184.983 460;P为样品测定次数,P=7;n为拟合工作曲线时的测定次数,n=1;C为预评估时由线性方程计算所得浓度,C=0.000 897 mg·L-1;Ci为标准工作溶液浓度,mg·L-1;为标准工作溶液浓度的平均值,mg·L-1。
经计算,样品测定引入的标准不确定度u(B5)为0.000 024 7 mg·L-1,因此校准曲线引入的相对标准不确定度为
2.4 合成标准不确定度
测定噻虫胺含量的相对合成不确定度为
合成标准不确定度u=X×ucrel=0.020 1×3.05%=0.000 61 mg·kg-1,扩展不确定度为U=k×u=2×0.000 61=0.001 2 mg·kg-1,试样中噻虫胺含量的检测结果报告为X=(0.020 1±0.001 2)mg·kg-1,k=2。
3 结论
通过对猪肉中噻虫胺残留量测定过程进行不确定度分析,得出试样中噻虫胺含量的检测结果为(0.020 1±0.001 2)mg·kg-1,k=2。在各不确定度分量中,标准曲线拟合对噻虫胺含量测定的不确定度影响较大。实验室要加强检测人员培训,提高检测人员的技术水平,在样品处理和检测过程中,严格按照标准方法进行操作,定期维护仪器,保证仪器的稳定性,减少检测过程中的不确定度。
