大肠杆菌必需基因的CRISPR-Cas9敲除策略及高效质粒置换方法
2023-10-09刘锦辉黄建峰尤晓颜张燕飞
刘锦辉,黄建峰,尤晓颜*,张燕飞*
1(河南科技大学 食品与生物工程学院,河南 洛阳,471023)2(中国科学院天津工业生物技术研究所,国家合成生物学技术创新中心,天津,300308)
必需基因通常被定义为无法从基因组删除的基因[1-2]。在原核生物如大肠杆菌中,必需基因对细胞基本功能的维持有着极为重要的作用,其对细胞存活是绝对必要的[3]。目前,尽管使用单基因删除、基于CRISPR/dCas系统的多基因抑制以及转座子诱变等方法可有效确定基因组必需基因,但是针对必需基因功能研究方法较少[3]。如何保证必需基因被敲除后细胞可保持正常生长,并探究必需基因突变体的正常功能表征是必需基因功能研究面临的难题,缺乏有效的遗传操作工具是造成这一困难的主要原因。
LIANG等[4]利用Flp/FRT系统并使用基因tet和sacB作为正反筛标记构建了一种可删除大肠杆菌基因组必需基因的方法。但是当目标必需基因发生突变或被改造而严重影响细胞正常生理功能时,可能引发逃逸现象而导致编辑效率显著下降。FAN等[5]报道了一种质粒改组的酵母必需基因编辑方法。将待编辑的必需基因yfeg克隆至回补质粒并使用URA3作为选择标记。在含有5-氟乳清酸的平板培养时,可筛选出无回补质粒的基因编辑菌株。该方法可以实现对必需基因的功能表征,但是选择标记筛选方法也可能使细胞发生突变进行逃逸。CRISPR/Cas9作为第三代基因编辑技术,自问世以来就广泛用于细胞基因组的编辑[6-7]。Cas9可在向导RNA(sgRNA)的靶向作用下被引导至目标基因靶点位置进行切割产生双链断裂,诱导细胞内源修复如非同源末端拼接或同源重组,即可实现基因组基因精准、高效且快速的删除、插入及替换操作[8-9]。目前,针对于大肠杆菌的CRISPR/Cas9基因编辑使用最多的是由JIANG等[10]开发的pCas/pTarget双质粒系统,该系统可实现大肠杆菌高效单基因编辑(效率高达100%)或多基因连续编辑,但是无法实现必需基因编辑。
为实现大肠杆菌基因组必需基因的高效、无痕编辑,我们以必需基因metK[11-13](编码的S-腺苷甲硫氨酸合酶催化L-甲硫氨酸与ATP反应生成生长必需因子S-腺苷甲硫氨酸)作为对象,构建了一种基于CRISPR/Cas9的基因组必需基因编辑方法。首先,构建携带有野生型metK基因但PAM位点被突变的编辑质粒pHL3。另外构建含有完整metK(PAM位点突变)表达盒的回补质粒。在消去pCas和pHL3之前转化回补质粒形成三质粒菌株,最终消去编辑质粒可获取含有回补质粒的基因组必需基因metK敲除菌株(图1)。探索了基于质粒不相容及Flp/FRT系统两种回补质粒置换方法,实现对回补质粒的高效置换,有助于MetK突变体的功能表征。通过生长表征,metK表达盒(野生型)克隆至质粒时对菌株生长无明显影响,而MetKK265A(对L-甲硫氨酸亲和力降低)突变体可使菌株生长与L-甲硫氨酸浓度相偶联。基于CRISPR/Cas9的必需基因编辑及质粒置换方法可实现高效基因组必需基因的无痕编辑,并对必需基因进行功能表征。所选取的MetKK265A突变体有望被开发作响应L-甲硫氨酸的生物传感器,实现L-甲硫氨酸高产菌的筛选。
1 材料与方法
1.1 试剂及仪器
DNA小量提取试剂盒、胶回收试剂盒,Omega公司;基因组DNA提取试剂盒,Promega公司;一步克隆试剂盒(One step clony Kit)、高保真DNA聚合酶,南京诺维赞有限公司;PCR清洁试剂盒,AxyGEN公司;DpnⅠ限制性内切酶,New England Biology公司;常规化学试剂,国药集团化学试剂有限公司;卡那霉素(Kanamycin,Kan)、大观霉素(spectinomycin,SD)、氯霉素(chloramphenicol,Cm),生工生物工程(上海)股份有限公司。
Eppendorf Eporator型电转化仪,德国Eppendorf公司;RePure-A型PCR仪,杭州柏恒科技有限公司;0.2 cm电击杯、ChemiDoc MP型凝胶成像仪,伯乐生命医学产品(上海)有限公司;MicroScreen HT型高通量微生物生长曲线分析系统,杰灵仪器制造(天津)有限公司。
1.2 菌株、质粒及相关引物
本研究中所使用或构建的菌株及质粒见表1。所使用引物均由北京擎科生物科技有限公司合成,序列详见表2。

表1 研究所使用菌株和质粒Table 1 Strains and plasmids were used in this study
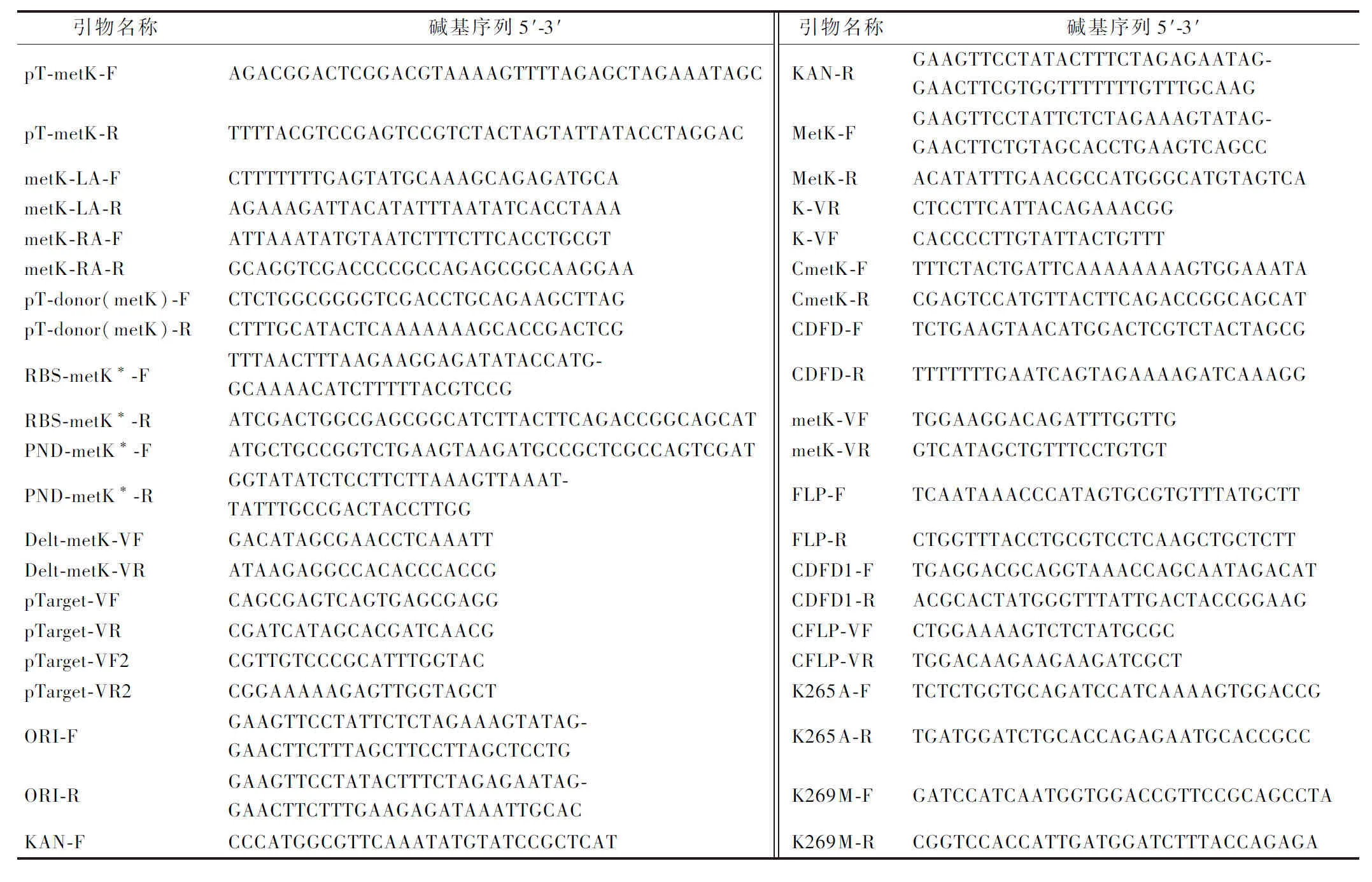
表2 研究所使用引物Table 2 Primers were used in this study
1.3 质粒构建
1.3.1 基因编辑质粒构建
以pTargetF质粒为模板,引物pT-metK-F/R突变N20序列,PCR结束后使用DpnⅠ消化模板,转化至DH5α感受态细胞,SD平板筛选转化子,pTarget-VF测序验证,构建质粒pHL1。以MG1655基因组为模板,引物metK-LA-F/R扩增500 bp左同源臂片段,引物metK-RA-F/R扩增500 bp右同源臂片段;以pHL1为模板,引物pT-donor(metK)-F/R扩增连接载体,三片段连接后转化至DH5α感受态细胞,SD平板筛选及pTarget-VF/VR测序验证,构建质粒pHL2。以质粒pHL2为模板,引物PND-metK*-F/R扩增载体片段;以MG1655基因组为模板,引物RBS-metK*-F/R扩增metK基因ATG到TAA部分的片段,大小约为1 155 bp。两片段消化、纯化后经One step clony Kit连接,转化后SD平板筛选,pTarget-VF2/VR2测序验证,构建编辑质粒pHL3。
1.3.2 回补质粒构建
以质粒pET28a(+)为模板,引物Kan-F/ R扩增Kan抗性筛选标记片段;以质粒pHL4为模板,引物ORI-F/ORI-R扩增复制子CloDF13片段,引物MetK-F/MetK-R扩增野生型metK表达盒片段,3个片段经消化纯化后连接并转化至E.coliDH5α感受态细胞中,Kan抗性平板筛选转化子并进行鉴定及测序,测序引物为K-VF/VR,获取回补质粒pHL8。
1.3.3 置换质粒构建
以E.coliMG1655基因组为模板,引物CmetK-F/CmetK-R扩增带有野生型启动子的metK基因表达盒,质粒pJH1为模板,引物CDFD-F/CDFD-R扩增质粒骨架,两片段经消化纯化后连接得置换质粒pHL9。以质粒pJH2为模板,引物FLP-F/FLP-R扩增flp片段,以pHL9为模板,引物CDFD1-F/CDFD1-R扩增连接的质粒骨架片段,两片段经消化纯化后连接得置换质粒pHL10。
1.4 必需基因metK编辑过程
参考JIANG等[10]报道,构建E.coliMG1655/pCas并制备为电转化感受态细胞,将编辑质粒pHL3(约500 ng)与E.coliMG1655/pCas电转化感受态细胞混匀,转移至0.2 cm电击杯中,调节电压至2.5 kV进行电击,电击完成后加900 μL LB培养基,220 r/min,30 ℃孵育2.5 h。孵育完成后4 500 r/min离心浓缩菌体,涂布至含有SD及Kan的双抗性平板,30 ℃倒置培养12~16 h。用引物Delt-metK-VF/VR进行菌落PCR,筛选基因组metK敲除的转化子。挑取metK敲除菌株,接种至含有SD和Kan抗性的LB培养基中,随后将其制备为化学感受态细胞并转化回补质粒pHL7,于含有Kan、SD和Cm抗性的LB平板筛选含有三质粒的metK敲除菌株。随后参考JIANG等[10]的方法依次消去pCas和pHL3,最终获得基因组metK敲除菌株。测序验证引物为Delt-metK-VF/VR。
1.5 必需基因metK敲除菌株生长测试
将野生型MG1655及metK敲除菌株接种至 10 mL LB培养基中,培养12~16 h作种子液。2个菌株生长测试使用使用杰灵快速生长监测系统MicroScreen HT进行。对于LB培养基,直接按1%接种量接种至含有50 mL LB的摇瓶中,37 ℃,220 r/min培养,间隔1 h取样测定OD600值,绘制生长曲线。对于M9培养基(6.8 g/L Na2HPO4, 3.0 g/L KH2PO4, 0.5 g/L NaCl, 1.0 g/L NH4Cl, 0.24 g/L MgSO4, 11.1 mg/L CaCl2, 4 g/L菌落糖),取1 mL种子液,4 500 r/min离心2 min后弃上清液,加1 mL M9培养基重悬菌体,按1%接种量接种至装有1 mL M9培养基的48孔板中。随后调节培养条件为800 r/min,37 ℃,观察菌株生长曲线,当菌株进入平台期后停止仪器运行。绘制菌株生长曲线。LB培养基和M9培养基生长测试分别设置3个平行重复。
1.6 质粒置换方法
将菌株E.coliMG1655 ΔmetK/pHL8制备为化学感受态细胞,转化置换质粒pHL9或pHL10,复苏完成后4 500 r/min离心3 min,弃上清液,用M9培养基重悬菌体,接种至10 mL含有Sm抗性的M9培养基中,220 r/min,37 ℃振荡培养至菌体生长。将培养物稀释105倍,分别取100 μL涂布于Sm单抗性平板和Sm+Kan双抗性平板,37 ℃倒置过夜培养12~16 h后统计平板菌落数。从Sm抗性平板中任意挑取20个单克隆,于Kan抗性平板划线,验证Kan抗性。根据Kan抗性验证情况,计算回补质粒的未置换效率。回补质粒的置换效率计算如公式(1)所示:
(1)
2 结果与分析
2.1 E.coli MG1655基因组必需基因metK的高效无痕敲除
为实现基因组metK必需基因的编辑,在metK基因内部距离起始密码子<30 bp的范围内选取PAM位点,取其上游20 bp的序列作为N20,各取ATG前及TAA后500 bp作为同源臂,并克隆基因组metK构建编辑质粒pHL3(图1-a),使基因metK编辑成功后细胞可正常生长。为防止Cas9对编辑质粒pHL3上metK的切割,将pHL3中metK的PAM位点突变为PAMCCT→PAMTCT。同时,为保证编辑工具质粒消除后,被编辑菌株可正常生长,将E.coliMG1655基因组完整metK基因表达盒克隆至质粒,并将PAM位点进行突变(PAMCCT→PAMTCT,表示为PAMLess)构建回补质粒pHL7及pHL8。参考JIANG等[10]报道对基因组metK进行编辑,筛选出metK敲除菌株后将其制备为化学感受态细胞并转化回补质粒pHL7或pHL8,构建三质粒菌株E.coliMG1655 ΔmetK/pCas-pHL3-pHL7(或pHL8),随后分别消去编辑质粒pCas和pHL3,最终获得携带有metK表达盒回补质粒的基因组必需基因metK敲除菌株E.coliMG1655 ΔmetK/pHL7及E.coliMG1655 ΔmetK/pHL8,命名为JHb1和JHb2(图1-c)。
根据验证引物在基因组位置,若metK未编辑成功则PCR扩增目的条带约为3.0 kbp,若编辑成功则目的条带大小为1.5 kbp。经PCR和核酸电泳验证,所挑取平板单菌落均为metK敲除(图2-a),成功获得metK敲除菌株E.coliMG1655 ΔmetK/pCas-pHL3。为验证基因组必需基因metK成功敲除,通过对PCR产物进行测序,结果表明基因组metK被敲除,符合PCR验证结果(图2-b)。
2.2 基于质粒不相容的质粒置换方法
为实现质粒置换,方便研究具有不同酶学性质的MetK突变体对细胞生长和生理影响,探索了高效质粒置换方法。首先采取质粒不相容原理策略,置换质粒与回补质粒含有相同的复制子[14]。当置换质粒进入细胞后,在Sm抗性选择压力下,回补质粒可被置换(图3)。为验证MetK为野生型或为突变体时质粒置换效率,除了选择野生型MetK,还有MetKK265A及MetKK269M[对L-甲硫氨酸的Km值分别为0.54 mmol/L和7.1 mmol/L,高于MetKWT(0.08 mmol/L),可能影响SAM的正常合成而影响细胞生长][15]2种突变体进行试验。结果见表3,在质粒不相容的选择压力下,当MetK为野生型时质粒置换效率可达(74.83±0.12)%,但是含有MetKK265A及MetKK269M的置换效率为(0±0)%。以上结果表明,对于Km值(对底物L-甲硫氨酸)变大的MetK突变体,仅依靠质粒不相容无法实现质粒置换,从而影响对该突变体的功能表征。

图3 基于质粒不相容的质粒置换方法Fig.3 Method of plasmid replacement based on plasmid incompatibility

表3 不同置换方法中回补质粒的置换效率Table 3 Replacement efficiency of the complement plasmid in different methods
2.3 基于Flp/FRT系统的质粒置换方法
为提高质粒置换效率,在质粒不相容的方法基础上,我们探索了第二种基于Flp/FRT位点特异性重组系统的质粒置换方法。在回补质粒复制子两端加入同向FRT位点构建回补质粒pHL8,并在置换质粒中加入flp基因构建pHL10。当pHL10进入细胞后,flp表达重组酶Flp识别FRT位点,使回补质粒分裂为两部分,一部分含有复制子但无metK及筛选标记,一部分有metK及筛选标记但无法复制(图4),此时宿主细胞无法保留野生型MetK,应该有助于提高质粒置换效率。基于此,我们重新验证该系统中,携带MetKWT、MetKK265A及MetKK269M置换质粒的置换效率。将pHL10转化至E.coliMG1655 ΔmetK/pHL8,在Sm抗性的选择压力下于M9培养基中培养,将培养物稀释涂布分离单菌落并验证Kan抗性,若存在Kan抗性则说明回补质粒pHL8置换失败;若无Kan抗性则说明回补质粒置换成功。随后挑取无Kan抗性菌株提取质粒验证突变有无回复突变。如表3所示,携带MetKWT、MetKK265A及MetKK269M置换质粒的置换效率可分别提升至(100±0)%、(100±0)%及(84.10±5.16)%。以上结果表明利用Flp/FRT系统可有效提高回补质粒的置换效率,促进对必需基因功能的研究。
2.4 基因组metK敲除菌株生长测试
为探索metK克隆至质粒后对细胞生长是否有影响,以野生型E.coliMG1655菌株作为对照,以编辑菌株JHb1(带有回补质粒pHL7)、JHb2(带有回补质粒pHL8)和JHb3(带有置换质粒pHL10,野生型metK表达盒)作为研究对象,选用LB培养基和以葡萄糖为唯一碳源的M9培养基进行生长测试。结果表明,在LB培养基中,编辑菌株与对照菌株保持相同生长趋势(图5-a)。但在M9培养基中对数生长期相对平缓,到达平台期时间延长,这可能是由于M9培养基仅含有葡萄糖作为碳源,缺乏其他生长因子所导致(图5-b)。说明带有野生型metK或PAM位点突变的metK克隆至质粒进行表达也可维持细胞正常生理活动。
本文还对携带MetKK265A的菌株在M9培养基进行生长监测。当培养基中仅以葡萄糖作为唯一碳源时,菌株生长受到严重抑制,培养30 h后OD600值仅为0.51,这可能是由于MetKK265A对L-甲硫氨酸的亲和力严重下降导致了SAM合成受限,进而影响了细胞正常生理功能。当培养基中添加有0.8 g/LL-甲硫氨酸时生长恢复,OD600值为1.98,略低于野生型(图5-c)。
3 讨论
必需基因作为细胞生命活动保守的基本功能单位,其功能研究可有助于理解生命的起源和进化[1]。但是由于早期缺乏有效的编辑工具,难以实现对必需基因的功能表征。本研究中,我们开发了一种基于CRISPR/Cas9的大肠杆菌基因组必需基因敲除方法,可实现基因组必需基因的高效无痕删除。在pCas/pTarget系统的基础上,将必需基因metK克隆至质粒pTarget构建编辑质粒pHL3以保证基因组metK成功编辑后细胞存活(图1-a),构建带有PAM位点突变的metK的回补质粒保证编辑质粒消去后细胞生长,经过两步电转及一步化学转化的方式可实现必需基因metK的高效删除(图1-c)。在前期构建时,PAM位点选取自metK基因中部,构建的基因编辑质粒pHL3经电转化筛选时,挑取大量单克隆进行PCR验证难以获取阳性克隆。经测序后发现,一部分基因组中PAM位点与编辑质粒中的PAM突变位点发生了重组,即基因组中PAMCCT→PAMTCT,一部分发生逃逸未实现编辑(图1-a)。根据λ-Red重组系统原理,当上下游修复模板长度>35 bp时即可实现高效的重组[16]。当PAM位点在基因中部时,Cas9切割后,pHL3中PAMTCT两端的序列可作为同源修复模板,导致metK编辑效率低下。为提高编辑效率,我们在据metK内部距ATG或TAA<30 bp的位置选取PAM位点,重新构建编辑质粒pHL3。经验证,所挑取的平板单克隆均为metK敲除菌株, 实现metK的100%编辑(图1-b)。
为实现对MetK突变体的功能表征,我们探索了质粒不相容和基于Flp/FRT系统的两种回补质粒置换方式。依赖于质粒不相容原理,当MetK为野生型时可实现(74.83±0.12)%的置换效率,但是对L-甲硫氨酸亲和力下降的突变体MetKK265A及MetKK269M无法实现质粒置换,这可能是因为突变体对L-甲硫氨酸亲和力下降,影响SAM合成效率,使底盘细胞通过自适应调节保留了回补质粒以维持细胞生长。在质粒不相容基础上加入Flp/FRT系统后,携带MetKWT、MetKK265A及MetKK269M的置换质粒对回补质粒的置换效率可达100%、100%及84.10%,说明基于此方式可有效实现回补质粒的高效置换。值得注意的是,MetKK269M虽然成功置换回补质粒,但是所获得菌株中MetKK269M均发生回复突变MetKM269K,说明当MetK突变体活力严重下降时,即使能够成功置换回补质粒细胞也会发生逃逸。这一现象说明基于Flp/FRT的质粒置换系统在解决回复突变问题上有待进一步改进。
经过LB培养基和M9培养基培养生长监测,质粒表达metK(野生型和含有PAM突变)的编辑菌株与野生型菌株E.coliMG1655无明显生长差异,说明基于质粒表达的metK可正常维持细胞生长。通过对突变体MetKK265A的生长表征,使用仅含有无机盐和葡萄糖的M9培养基培养时细胞生长会受到严重限制,但是这一限制可以通过外源L-甲硫氨酸的添加恢复,这一表征结果说明基于Flp/FRT的质粒置换系统对必需基因功能表征的可行性。另外,突变体MetKK265A的生长表征结果使得其有望被开发作为可响应L-甲硫氨酸的生物传感器,用作L-甲硫氨酸高产菌株的高通量筛选。
