BRAF 基因变异8 例临床分析
2023-09-20欧跃徐段远辉李洁玲
欧跃徐 段远辉 曹 洁 李洁玲
重庆医科大学附属儿童医院全科医学科 儿科学重庆市实验室 儿童发育疾病研究教育部重点实验室 儿童发育重大疾病国家国际科技合作基地 国家儿童健康与疾病临床医学研究中心(重庆 400014)
BRAF基因位于7号染色体长臂3区4带(7q34),其编码的B-Raf 蛋白属于生长信号转导蛋白激酶的Raf激酶家族成员,它涉及多种信号转导途径,主要调节RAS/MAPK/ERKs 信号通路,从而影响细胞分裂、分化和分泌,调控炎症、凋亡、癌化及肿瘤细胞的侵袭和转移[1]。BRAF基因变异会使RAS/MAPK/ERKS 通路被过度激活,出现与发育障碍相关的RAS 通路病以及各种肿瘤性疾病[2]。其中源于生殖细胞的BRAF基因变异导致的RAS 通路病包括心-面-皮肤综合征(cardio-facio-cutaneous syndrome,CFCS)、努南综合征(Noonan syndrome,NS)、努南综合征伴多发雀斑(Noonan syndrome with multiple lentigines,NSML)等,而根据BRAF基因变异的位点不同,临床表现也会有一定的差异和重叠[3]。此外,BRAF基因变异在体细胞中可出现朗格罕斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)、黑色素瘤及其他肿瘤疾病,而BRAF600E位点的错义变异c.1799T>A(p.V600E)是其最常见的变异方式[4]。本研究对重庆儿童医院诊治的8例存在BRAF基因变异患儿的临床表现、治疗情况及预后进行回顾性分析,并分析其临床表型与基因型的相关性。
1 资料与方法
1.1 临床资料收集
收集2015年1月至2022年3月就诊,静脉血或病理组织基因测序显示存在BRAF基因变异的8 例患儿的临床资料,包括就诊年龄、性别、出生史、生长发育史、智力运动水平、临床表现、治疗及预后情况、影像学结果、实验室检测结果、病理结果、基因检测结果等。本研究经医院医学伦理委员会批准(No.2022年伦审(研)第475号)及所有患儿监护人的知情同意。
1.2 方法
1.2.1 基因检测 4 例患儿因存在发育落后和特殊面容,对患儿及其父母各抽取静脉血2 mL进行全外显子组(WES)基因检测;另外4 例既往体健、发育正常的患儿,因发现体内包块(纵膈、枕部、颅内)经外科手术切除包块后送病理活检及基因检测,采用二代高通量测序基因芯片捕获技术进行基因检测,再根据基因检测结果对致病变异以Sanger测序验证。
1.2.2 实验室检查 所有患儿均进行三大常规、电解质、肝肾功能、心肌酶谱等检查,部分患儿根据症状选择性行彩色多普勒超声心动图、心电图、头颅核磁共振、脑电图、CT、Gesess发育评估等辅助检查,了解病情并排除其他病因。
2 结果
2.1 基因和病理结果
存在发育落后和特殊面容的4 例患儿,均为起源于生殖细胞7号染色体上的BRAF基因新发变异,根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南致病等级均提示致病,父母均为野生型,但基因变异位点各有不同(表1)。发现体内包块的4例患儿,病理活检结果为LCH 2例、额叶胶质神经元混合性肿瘤1例、小脑毛细胞星形细胞瘤1例,基因检测(WES)结果均为来源于体细胞的BRAF600 E 位点的变异。

表1 4例CFCS患儿临床表现
2.2 临床资料
2.2.1 源于生殖细胞变异的4例CFCS患儿临床表现 起源于生殖细胞BRAF基因变异的4 例患儿中男2例、女2例,就诊年龄4月13天~2岁,就诊原因包括发育迟滞2例,频繁惊厥发作2例。2例因胎膜早破而早产,2例为足月儿但出生时有轻度窒息史,母孕史均正常,无家族史。均存在特殊面容,4 例有眼距宽、鼻梁低、耳位低,3例有小下颌、人中沟深,2例有耳廓畸形、额头高大、睑裂外斜;全部存在皮肤及毛发改变,表现为皮肤干燥粗糙,掌纹深,眉毛秃脱,在秃顶的区域存在稀疏、卷曲、脆的头发(图1)。3例存在心脏结构异常,包括小型房间隔缺损(3例)、肺动脉狭窄(1例)、室间隔及左室壁阶段性增厚(1例)。心电图均无异常。4例患儿均存在严重的神经心理表型,包括智力运动发育迟滞(4例)、癫痫(2例)、临床下放电(2例)等,同时还存在身材矮小(1例)、喂养困难(1例)等。除以上核心症状外,还存在其他系统异常,包括甲状腺功能减退、血管瘤、先天性喉软骨发育不良、隐睾、斜视、睡眠障碍等。见表1。
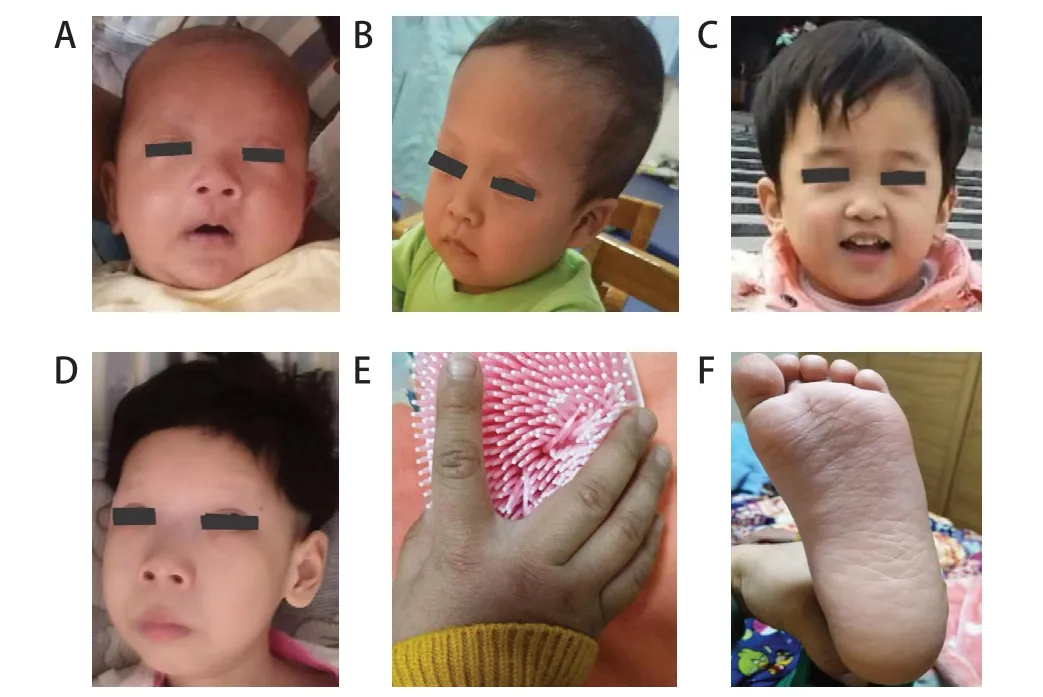
图1 4 例CFCS 患儿的面部特征及皮肤改变
4例CFCS患儿在初次就诊时均进行了Gesell评估,结果显示其四大能区DQ值均<65分,均表现为智力运动发育迟滞(表1)。身高体重评估显示例2身高<-3SD,体重在-3SD~-2SD,表现为身材矮小;例4 因喂养困难,体重<-3 SD,身高在中位数,表现为重度营养不良。4例患儿中2例存在癫痫。其中例4为难治性癫痫,初为有热惊厥,后演变为无热惊厥,发作形式为局灶继发全面性发作,表现为右侧嘴角及右侧肢体抽动继发全身强直阵挛发作,每次持续1~3 min,10~20次/d;脑电图示全脑多灶性高幅不规则棘/尖波、棘/尖-慢波发放,醒睡各期均呈电持续状态;头颅MRI 示脑萎缩,脑室旁白质发育不良,幕上脑室明显扩张;先后经丙戊酸钠、左乙拉西坦、奥卡西平、硝西泮、生酮饮食、妥泰等治疗不能完全控制,现仍有发作,约2次/d,呈癫痫脑病状态,长期卧床,重度营养不良,生存质量极差。例2为无热惊厥,表现为全面性强直阵挛发作,3~12次/d,每次持续2~3 min;脑电图示睡眠期两侧额中央区为主3~4 Hz棘/尖-慢波、尖波同步/不同步发放;头颅MRI 示胼胝体偏薄,予左乙拉西坦治疗后未再发作,但脑电图无明显改善。例1和例3虽无惊厥发作,但存在临床下放电,脑电图均示睡眠期多见顶区/额区3~4 Hz尖-慢波同步/不同步发放伴扩散,均存在睡眠障碍,表现为睡前兴奋、入睡难、睡眠时间减少、夜间哭闹等;例3头颅MRI示脑萎缩样改变,胼胝体较薄;先后予丙戊酸钠、左乙拉西坦治疗,睡眠障碍稍有改善,脑电图无明显改善。例1头颅MRI提示髓鞘化延迟,未予抗癫痫药物治疗,睡眠障碍及脑电图均无明显改善。
4例患儿均存在心脏-特殊面容-皮肤毛发改变-神经系统异常表现,结合基因检测均有BRAF基因变异,故均诊断为心-面-皮肤综合征(CFCS)。
2.2.2 源于体细胞BRAF600E变异的4例患儿临床表现 4 例患儿中男3 例、女1 例,就诊年龄1 岁1月~13岁,就诊原因包括发现纵膈肿块1例、枕部包块1 例、颅内肿块2 例,其中2 例为LCH,2 例为神经系统恶性肿瘤。1 例LCH 患儿发热1 个月,胸部增强CT 示前上纵膈占位,伴肿块内钙化;另1 例为发现枕部包块3 个月,烦渴、多饮多尿2 个月,头颅增强CT 示左侧颞骨、眼眶外后壁、蝶骨翼骨骨质破坏伴局部软组织肿块影,2 例患儿均行肿块切除后再行LCH 方案化疗及达拉菲尼靶向治疗。2例神经系统肿瘤患儿中,1 例表现为头痛、呕吐伴双下肢乏力2 个月,头颅MRI 示右额叶巨大占位性病变(56.8 mm×81.7 mm),手术切除后1年出现视物模糊,复查头颅CT 示右侧额叶异常强化影(4 mm×4 mm×3.5 mm),考虑肿瘤复发再次行手术切除,病理检查示右侧额叶胶质神经元混合性肿瘤;另1 例表现为阵发性头痛2 周,头颅CT 示后颅窝占位性病变伴梗阻性脑积水,手术切除后病理检查示右侧小脑毛细胞星形细胞瘤。
4 例患儿切除的肿块送基因检测均提示为BRAF600 E 基因变异。患儿目前均未复发,可正常生活。
3 讨论
BRAF基因包含18 个外显子,编码651 个氨基酸,目前已有超过82种致病变异[5]。BRAF基因编码的RAF家族丝氨酸/苏氨酸蛋白在调控RAS/MAPK信号通路中起关键性作用,在该通路中RAF 蛋白激酶作为RAS 蛋白下游的效应蛋白激活MEK 蛋白,MEK 蛋白激酶再激活ERK 蛋白,ERK 蛋白激酶最后作用于下游的效应蛋白,形成RAS-RAF-MEKERK 信号通路,影响细胞的增值、分化、衰老、凋亡等[6]。BRAF基因变异则会导致RAS/RAF/MEK/ERK级联中的信号转导失调,导致自发地向下游激酶发出信号,引起MEK 蛋白激酶和ERK 蛋白激酶对下游信号的过度激活,最终导致细胞生长、增值和分化异常,从而产生与发育障碍相关的RAS 通路病以及各种肿瘤性疾病[7]。
RAS 通路病是指一组以RAS/MAPK 通路异常为共同发病机制的遗传性疾病,除BRAF基因外,其他影响该通路的基因变异也会导致RAS 通路病,如MEK1、MEK2、KRAS、PTPN11、RAF1、NF1、HRAS等基因。它包括7种亚型:CFCS、NS、NSML、I型神经纤维瘤病(neurofibromatosis type 1,NF1)、Costello综合征(Costello syndrome,CS)、Legius综合征(Legius syndrome,LS)、毛细血管畸形-动静脉畸形综合征(capillary malformation-arteriovenous malformation syndrome,CM-AVM)[8]。RAS通路病的总发病率为1/1000,其中NS最常见,发病率约1/2500~1/1000,其次是NF1,发病率约1/3000,CS和CFCS非常罕见,发病率分别为1/290000和1/810000[5]。由于它们具有共同的发病机制,故临床表现、基因型-临床表型的对应关系均存在部分交叉重合。BRAF基因变异导致的RAS 通路病包括CFCS、NS、NSML 这3类,而在CFCS患儿中存在4种基因变异类型(BRAF、MAP2K1/MAP2K2、KRAS),其中BRAF基因变异占75%,MAP2K1/MAP2K2占25%,KRAS占2%~3%[9]。
CFCS是一种罕见的常染色体显性遗传病,核心症状包括特殊面容、心脏畸形、皮肤毛发病变及神经系统异常等,其他还可能出现喂养困难、肌张力低下、隐睾、斜视、眼球震颤、听力异常、身材矮小等表现;特殊面容常表现为相对大头畸形、额头高大、双颞变窄、睑裂下斜、低鼻梁、人中沟深、小下颌、耳位低、耳轮后旋等,心脏畸形包括肺动脉瓣狭窄、肥厚性心肌病、房间隔缺损、室间隔缺损、心律失常等,皮肤毛发病变表现为干燥症、鱼鳞病、血管瘤、皮肤过度角化、毛发卷曲/稀少/易断、眉毛及睫毛稀少或缺如等,神经系统异常包括智力运动发育迟滞、癫痫、脑病等[10]。
本文4 例CFCS 患儿均源于生殖细胞BRAF基因变异,变异位点分别为exon12、exon15、exon6、exon16,这些位点既往已有报道,且均为致病变异[11]。4 例患儿均存在CFCS 的核心症状,其中1 例无心脏畸形,其他3例虽有心脏结构畸形,但主要表现为小型房间隔缺损,1 例伴肺动脉狭窄,1 例伴室间隔及左室壁增厚表现,均无心律失常表现,并且随着年龄增长自行恢复正常,这与文献报道的CFCS 患儿心脏畸形以肺动脉狭窄、肥厚性心肌病最多见不符[12]。目前并没有文献报道其心脏畸形的预后情况。4例患儿最主要、最严重的表现为神经系统异常,均存在严重的智力运动发育迟滞,脑电图均有明显痫性放电且头颅MRI也存在结构异常。其中例2、例4有癫痫发作,例2予以左乙拉西坦治疗后未再发作,但脑电图及智力改善不明显;例4表现为难治性癫痫、癫痫脑病,抗癫痫药物治疗效果差,智力极差。这与文献报道的CFCS患儿90%存在智力障碍,50%以上存在癫痫和脑病,且更多的是难治性癫痫相符[13]。例1、例3有脑电图异常,但无癫痫发作,表现为临床下放电,其脑电图为睡眠期放电明显且存在睡眠障碍,例3加用丙戊酸钠、左乙拉西坦治疗,睡眠障碍有减轻,但脑电图及智力改善不明显;例1未使用抗癫痫药物(AEDs),其睡眠障碍、脑电图、智力均无改善。目前尚没有文献报道CFCS 患儿临床下放电及睡眠障碍情况。根据4例患儿的临床表现应与NS相鉴别,由于具有共同的发病机制,NS 的临床表现与CFCS有很多交叉重叠。CFCS 和NS 均可出现相同的核心症状,但也有一些鉴别点,NS 多伴有隐睾和身材矮小,有“男性中的Turner”之称,且血液系统异常较为常见,此外NS患儿中50%以上均为PTPN11基因变异[14];而CFCS 以皮肤毛发特征性改变及神经系统异常等外胚层异常为主要表现,且在CFCS 患儿中75%为BRAF基因变异。本文4例患儿均为BRAF变异,且均以神经系统异常为最突出的表现,均存在特征性的皮肤毛发改变,故更符合CFC综合征的诊断。
此外,BRAF也是一个常见的原癌基因,当它在体细胞变异时可引起各种肿瘤性疾病,包括黑色素瘤、LCH、甲状腺癌、肺癌、直肠癌等,据报道,67%黑色素瘤及30%其他肿瘤疾病存在BRAF变异,其中V600E是最常见的变异类型[2、15],本文4例体细胞BARF变异患儿均为该类型。4 例患儿中,2 例为LCH,2例为颅内肿瘤,均采取手术切除加化疗的治疗方案,其中2 例LCH 加用了BRAF抑制剂——达拉菲尼靶向治疗;2 例颅内肿瘤分别为额叶胶质神经元混合性肿瘤和小脑毛细胞星形细胞瘤,目前均未复发。
综上所述,BRAF基因变异在生殖细胞时会导致RAS 通路病,以CFCS 常见,其存在多系统病变,以神经系统异常为突出表现,预后差,临床上应提高对这类疾病的认识,尽早完善基因检测,目前对于这类罕见遗传病尚无有效治疗,多以对症支持、康复、控制癫痫为主,应加强产前检查手段,减少该病胎儿的出生率。此外,体细胞BRAF基因变异还与肿瘤的发生及LCH 等相关,及时完善基因检测,早期进行手术切除,采取针对性的化疗方案及靶向治疗,可减少复发率,提高生存率。
