儿童自身免疫性胶质纤维酸性蛋白星形胶质细胞病临床分析
2023-09-20陈文雄廖寅婷吴文晓朱海霞彭炳蔚曾意茹吴汶霖陈宗宗李小晶
侯 池 陈文雄 廖寅婷 吴文晓 田 杨 朱海霞 彭炳蔚 曾意茹 吴汶霖 陈宗宗 李小晶
广州市妇女儿童医疗中心神经内科(广东广州 510120)
脑炎或脑膜脑炎是儿童神经系统常见的疾病之一,其中部分病例具有免疫介导的病因,类固醇激素治疗有效[1]。2016年梅奥医学中心描述了一种新形式的脑膜脑炎,称为自身免疫性胶质纤维酸性蛋白星形胶质细胞病(autoimmune glial fibrillary acidic protein astrocytopathy,GFAP-A)[2],随后在国际上得到证实[3-4]。该病的发现对部分不明原因的脑炎或脑膜脑炎的诊治有重要意义。目前对于该病的报道主要为成年患者,对儿童病例的研究不多,本研究系统报道了广东地区儿童病例的临床特点、治疗和预后。
1 对象与方法
1.1 研究对象
回顾性分析2018年6月至2022年7月在广州市妇女儿童医疗中心神经内科诊断为GFAP-A[2-4]患儿的临床资料。结合既往的研究[2-4],本研究的纳入标准:①年龄<18岁;②临床上存在不明原因发热、头痛和/或神经系统受损的症状及体征;③使用转染细胞-间接免疫荧光法检测出脑脊液胶质纤维酸性蛋白(GFAP)-IgG阳性。排除标准:感染、中毒、代谢、肿瘤等因素所致的神经系统病变。
本研究获得医院医学伦理委员会批准(穗妇儿伦审批第2016061601号)。患儿的监护人均签署知情同意书。
1.2 方法
1.2.1 临床资料 收集患儿的临床资料,实验室、影像学资料、治疗及随访资料。病程中神经功能的残障程度采用扩展致残量表(expanded disability status scale,EDSS)评估。通过神经内科门诊定期复诊问诊,神经系统查体,复查相关的实验室及影像学指标获得患儿的随访资料,随访周期为出院后1周、2周、1个月,随后每月定期门诊复诊。
1.2.2 脑脊液抗GFAP抗体检测 使用转染细胞-间接免疫荧光测定法在大鼠海马和小脑组织上测试抗GFAP 抗体。未稀释的脑脊液在室温下与载玻片上的组织切片反应3 小时,然后将载玻片用磷酸盐缓冲液冲洗2 次,再与荧光素偶联的山羊抗人IgG孵育2 小时。最后用磷酸盐缓冲液冲洗载玻片,并在显微镜下检查荧光强度。使用转染了GFAP α和GFAP ε 基因的HEK 293 细胞,通过基于细胞的测定法对所有样品进行了重新测试。脑脊液未经稀释制备,样品在室温下与GFAP 转染的细胞反应60 分钟。将载玻片用磷酸盐缓冲液冲洗3 次,然后与荧光缀合的山羊抗人IgG 孵育60 分钟。最后用磷酸盐缓冲液冲洗载玻片,并在显微镜下检查荧光强度。
1.2.3 治疗方案 急性期首先给予静脉甲基泼尼松龙冲击(intravenous methylprednisolone,IVMP)联合静脉滴注人免疫球蛋白(intravenous immunoglobulin,IVIG)治疗。IVMP治疗方案为10~15 mg/(kg·d)起始,其后每隔3~5天对半逐渐递减至2 mg/(kg·d)时,改为口服泼尼松龙维持。IVIG总量为2 g/kg,分3~5天输注完成。
1.3 统计学分析
采用SPSS 23.0软件进行统计分析。计量资料符合正态分布的以均数±标准差表示,非正态分布的以中位数(最小值~最大值)表示。计数资料以例数(百分比)表示。以P<0.05为差异有统计学意义。
2 结果
2.1 一般临床资料
本研究5例患儿祖籍均为中国广东,男3例、女2例,中位起病年龄为11.0(5.0~13.5)岁。其中1例患儿起病前2 年在外院诊断孤独症,目前处于康复训练过程中,表现为可完成简单指令,会说简单字词,不会说短语,EDSS 评分为3.5 分。其余患儿起病前均体健。
3例患儿在起病前存在前驱事件。1例在起病前1周患急性上呼吸道感染,对症处理后治愈;1例在起病前1 周患荨麻疹,口服组胺类药物抗过敏后皮疹消失;1 例在起病前2 周有接种疫苗。4 例首发症状为发热(热峰39.8~40.3 ℃),其神经系统首发症状包括头痛(3例)及共济失调(1例);1例首发症状为共济失调。急性期病程中出现的神经系统症状包括难以忍受的头痛(5例),意识水平下降(4例),共济失调(4例),肢体瘫痪(3例),睡眠障碍(3例),语言障碍(3例,其中1例起病前语言发育迟缓,起病后语言发育进一步倒退),自主神经功能障碍(3 例),抽搐发作(2例),精神行为异常(2例),眼球运动障碍(1例)。急性期神经系统阳性体征包括:颈抵抗伴克氏征阳性(5例),宽基底步态(3例),意向性震颤(2例),双下肢瘫痪(2例),腱反射减弱/消失(2例),眼外肌麻痹(1例),四肢肌张力增高(1例)。急性期EDSS评分为(7.0±1.4)分。患儿起病至确诊时间为(17.0±2.0)天。见表1。
2.2 一般实验室检查
病程中首次外周血常规白细胞计数(WBC)为(9.9±2.6)×109/L,中性分类为(51.9±6.8)%,血红蛋白、血小板、C-反应蛋白均正常,2 例患儿红细胞沉降率升高,其余均正常。中位血钠水平为126.4(118.7~138.3)mmol/L,4例患儿血钠低于正常值(血钠正常值范围135~145 mmol/L),血钾、血氯、血钙及血镁正常。
2.3 脑脊液改变
所有患儿在急性期均进行腰椎穿刺脑脊液检查,中位颅内压为230 mmH2O。1例患儿脑脊液改变为蛋白-细胞分离,脑脊液寡克隆区带蛋白阳性,葡萄糖及氯化物正常。其余4 例患儿脑脊液中位WBC为194.0(167.0~320.0)×106/L,淋巴分类为0.92(0.82~0.99),脑脊液蛋白为(1.2±0.4)g/L,脑脊液中位葡萄糖水平2.5(2.2~2.6)mmol/L(正常值范围2.8~4.5 mmol/L),3 例患儿脑脊液寡克隆区带蛋白阳性,脑脊液氯化物正常。所有患儿脑脊液GFAP 抗体阳性,中位抗体滴度为1∶10(1∶100~1∶10)。1 例患儿同期脑脊液抗N-甲基-D-天冬氨酸受体(N-methyl-D-aspartate receptor,NMDAR)抗体阳性,滴度为1∶10。
2.4 其他相关检查
所有患儿急性期行甲状腺功能检查,4例存在甲状腺功能异常,包括游离三碘甲状腺原氨酸(FT3)下降3 例,游离甲状腺素(FT4)升高1 例,未予特殊处理,1个月后复查均恢复正常。所有患儿抗甲状腺球蛋白及抗甲状腺过氧化物酶阳性。3例合并抗核抗体及抗干燥综合征抗体A阳性。4例患儿早期拟诊为颅内感染,病原学检查阳性,1例外周血肺炎支原体抗体阳性,1例外周血及脑脊液单纯疱疹病毒IgM阳性,1例脑脊液病原学宏基因组测序检出单纯疱疹病毒6型(序列数1),1例脑脊液病原学宏基因组测序检出肺炎链球菌及EB 病毒(序列数分别为15、4)。急性期3 例患儿脑电图异常,表现为脑电图背景活动慢。所有患儿视觉诱发电位及听性脑干诱发电位无异常。
2.5 影像学检查
所有患儿在急性期行MRI 检查,1 例表现为双侧侧脑室轻度扩张;其余4 例患儿MRI 改变包括双侧大脑半球柔脑膜强化(4例),基底节、放射冠多发异常信号(2例),脊髓软脊膜异常强化(2例),颈段脊髓见异常信号(1例)。见图1。
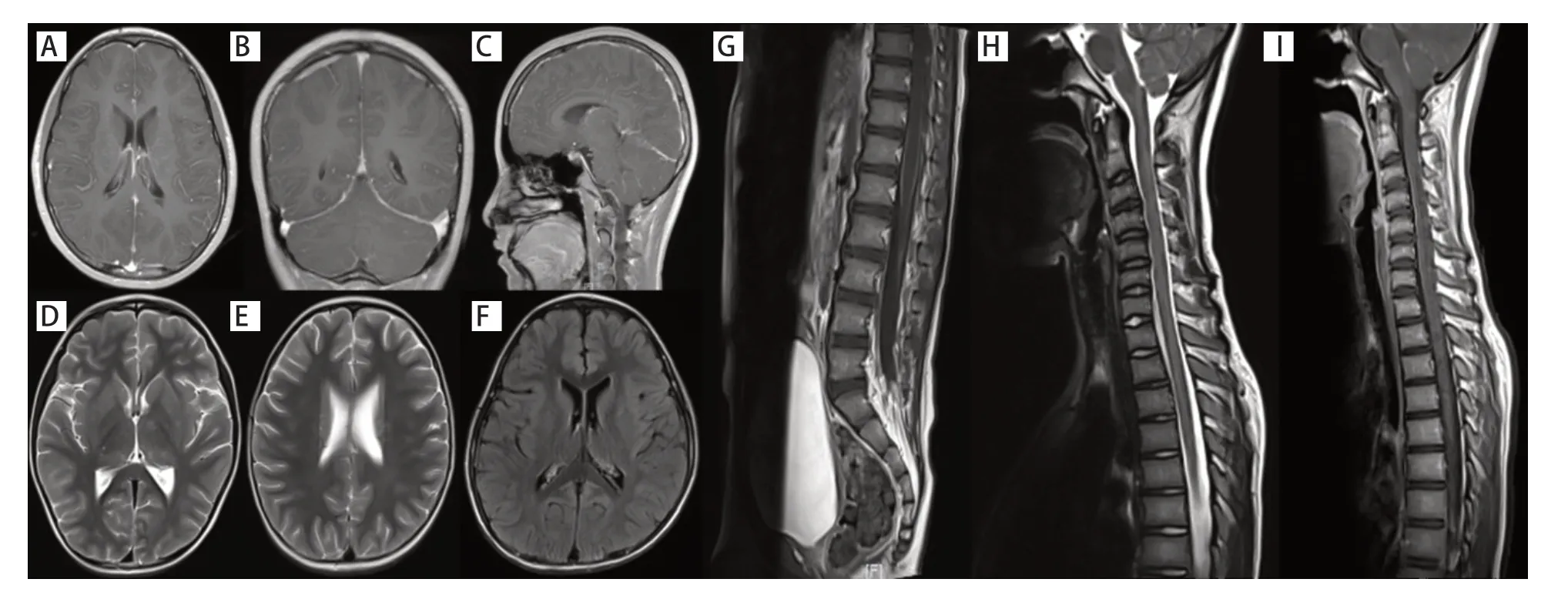
图1 GFAP-A 患儿MRI 表现
2.6 治疗
急性期所有患儿首先接受IVMP 联合IVIG 治疗。患儿从起病到开始接受IVIG治疗的中位时间为10.0(7.0~17.0)天,从起病到开始接受IVMP的中位时间为18.0(15.0~26.0)天。2例患儿经过1个疗程的IVIG联合IVMP治疗后缓解,2例患儿经过1个疗程IVMP及2个疗程的IVIG治疗后病情缓解,1例患儿合并脑脊液抗NMDAR 抗体阳性,经过2 个疗程IVMP 及IVIG 治疗后病情无缓解,给予利妥昔单抗(RTX)治疗后缓解。
急性期抗感染治疗包括静脉输注阿昔洛韦抗病毒(2例),口服阿奇霉素(1例),静脉输注美罗培南抗感染(1例)。急性期5例患儿均予静脉输注20.0%甘露醇及3.0%氯化钠降低颅内压,7.0(5.0~10.0)天后颈抵抗及克氏征消失。
急性期后所有患儿口服泼尼松龙维持治疗,1例患儿随访4个月暂未停药,其余4例患儿泼尼松龙维持治疗的中位时间为5.0(4.0~8.0)月。
2.7 预后随访
5 例患儿急性期出院至末次随访的中位时间为8.0(4.0~36.0)月。1例患儿末次随访4个月时,EDSS评分恢复到起病前的3.5分,复查MRI示颅内原有病灶吸收,见新发病灶,无相应的神经系统症状及体征,继续口服泼尼松龙及随访观察。其余4例患儿EDSS评分均为0分,复查MRI示病灶完全吸收。
3 讨论
GFAP在中枢神经系统表达,主要分布于星形胶质细胞中。最近提出了星形细胞病的新概念,包括视神经脊髓炎谱系障碍和GFAP-A[2,5]。与以水通道蛋白4 抗体为特征的视神经脊髓炎谱系障碍不同,GFAP-A是与IgG结合GFAP相关的脑膜脑脊髓炎。目前关于儿童病例的研究仍不多,本病例系统报道了中国广东地区儿童病例的临床特点。作为一种罕见病,该病相关的流行病学数据有限,可发生在任何年龄,疾病发生率没有显著的性别优势[3,6-8]。
该病多为急性或亚急性起病,病程为进行性加重或复发缓解模式,临床表现包括发热、头痛、脑病、运动障碍、视力异常、共济失调、精神障碍、癫痫及自主神经功能异常等[6-8]。脑膜脑脊髓炎或其部分表现形式是该病最常见的临床表型,儿童与成人临床表现相似[7-8]。与既往报道的儿童病例相似,发热、头痛及颈抵抗是最常见的症状及体征,大多数患者最初被怀疑患有感染性脑膜脑炎并接受了抗菌疗法治疗[7]。该研究中4例患儿早期拟诊为颅内感染,此外近年来也有单纯疱疹病毒脑炎后继发GFAP-A的报道[8-9]。因此推测在儿童GFAP-A中感染是很重要的诱发因素。本研究中1 例患儿临床表型为Miller-Fisher 综合征,表现为眼外肌麻痹、共济失调及腱反射消失三联征,脑脊液见蛋白-细胞分离。既往有报道GFAP-A表现为多发性周围神经炎症性脱髓鞘病[10-11],但Miller-Fisher综合征表型为本研究首次报道,进一步扩展了该病的表型。
与既往研究类似的是,在多数儿童GFAP-A 中脑脊液WBC及蛋白均升高[7-8]。此外本研究还发现患儿脑脊液葡萄糖水平有不同程度下降,这可能与该病累及脑膜致血脑屏障通透性改变有关。重叠抗体在GFAP-A中很常见,该病可共患NMADR抗体及抗髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体相关的自身免疫性脑炎或脱髓鞘脑病[7-8,12]。本研究中还发现多数患儿合并系统性自身抗体及甲状腺自身抗体阳性伴甲状腺功能一过性异常。因此,对于儿童GFAP-A尤其是临床表现不典型者需进行其他抗体相关的自身免疫性疾病的筛查。
该病中枢神经系统MRI 异常很常见,颅内病变可涉及皮质下白质、基底节、下丘脑、脑干、小脑及脑膜等[13]。本研究发现儿童病例最常见的病变为柔脑膜的强化,颅内病变虽然存在,但并未见到文献中描述的血管周围线性放射状强化的特征性病变及广泛的白质病变,这可能是儿童GFAP-A漏诊的原因。该病脊髓病变多为长节段性,本研究与既往研究类似的是,与水通道蛋白4 抗体相关的视神经脊髓炎谱系障碍比较,在GFAP-IgG阳性的脊髓炎中,脊髓中央管强化、脊髓实质点状强化或软脊膜强化是典型的MRI改变[14]。
该病目前暂无标准的治疗方法,急性期的治疗包括大剂量皮质类固醇,IVIG 和血浆置换。长期治疗包括口服皮质类固醇和免疫抑制剂[2-3,6,15]。与既往研究结果类似[8],本研究中多数患儿对皮质类固醇治疗反应良好,合并抗NMDAR 抗体阳性者对一线免疫治疗效果差。该病有一定的复发率,可在皮质类固醇激素减量的过程中或停用激素后复发,对于复发患儿,应考虑再次使用皮质类固醇激素并维持治疗3个月以上或使用免疫抑制剂治疗[15]。
综上,儿童GFAP-A在以发热、头痛为首发症状的脑膜脑脊髓炎中常见,早期需与颅内感染鉴别。儿童病例中柔脑膜强化是最常见的影像学改变。该病多数患儿一线免疫治疗有效,较少遗留神经系统后遗症。
