等离子体催化重整CH4/CO2中的等离子体强化表面反应动力学研究
2023-09-19孙进桃秦婉月
孙进桃,陈 琪,秦婉月
(北京交通大学 机械与电子控制工程学院,北京 100044)
甲烷(CH4)干式转化能够在减少温室气体排放的同时生成新的碳基能源和高附加值化学品。CH4/CO2重整反应具有强烈的吸热特性,只有反应温度≥645 ℃时在热力学上才是可行的[1]。传统的热催化虽然可以获得较高的转化活性,但是过高的反应温度会造成高能耗,而且积炭引起的活性中心的碳覆盖会导致催化剂活性降低甚至失活。非平衡等离子体含有大量的高能活性物质,能够有效改变低温反应路径,易于实现低温活化转化[2]。但非平衡等离子体中的电子处于不同的能量范围,放电过程中不可避免地生成副产物,目标选择性差。
通过非平衡等离子体与催化剂的耦合,形成一种新型强化反应手段,除了为反应过程提供催化剂表面路径,还提供了特有的气相路径。等离子体催化通过气相与催化剂表面这两条路径的协调,不仅可以降低催化反应温度,而且能够实现碳基燃料活化转化体系中的低温活化、低能耗、高收率及其高选择性[3],为解决温和条件下CH4/CO2转化这个前沿性科学问题,提供了一种极具发展前景的新方法。近年来,研究者们利用非平衡等离子体-催化剂协同方法开展了大量的活化转化CH4/CO2实验研究[4-5]。例如,等离子体-催化剂协同使得CH4/CO2单步合成高附加值的液体化工品变成可能[6]。但是,等离子体-催化剂协同作用中存在的若干复杂作用机理一直是以甲烷干式转化为代表的碳基燃料活化转化体系面临的难题,亟需推进和解决。
基于以上讨论,构建了包含等离子体反应机理、气相反应机理、表面反应机理的详细等离子体-催化协同CH4/CO2重整机理并开展零维动力学模拟;并结合NSD等离子体与Ni/SiO2催化剂协同CH4/CO2重整实验研究,为动力学模拟提供实验验证数据;通过表面反应路径和敏感性分析,研究等离子体中活性物质对表面反应动力学的强化效应。
1 实验和模拟方法
1.1 实验部分
1.1.1 催化剂的制备和表征
采用过量浸渍法,以氧化硅(20 nm,阿拉丁试剂,分析纯)和六水合硝酸镍(阿拉丁试剂,分析纯)制备质量分数为10%的Ni/SiO2催化剂(m(Ni)∶m(SiO2)=1∶9)。按照理论负载量称取相应质量的Ni(NO3)2·6H2O和SiO2加入到去离子水中配制成溶液。混合溶液在60 ℃恒温水浴中持续搅拌2 h,再在25 ℃下连续搅拌12 h,随后加热到85 ℃使水分蒸发。将黏稠状溶液放入烘箱在110 ℃下干燥5 h,然后在马弗炉中500 ℃煅烧5 h得到NiO/SiO2前驱体。将样品放置在管式炉中,通入100 mL/min的5%H2/95%N2混合气,在550 ℃下还原8 h。经压片、过筛制成粒径为40~60目的Ni/SiO2催化剂。采用XRD、XPS对制备的催化剂的晶体结构、元素价态等进行表征,结果如图1所示。
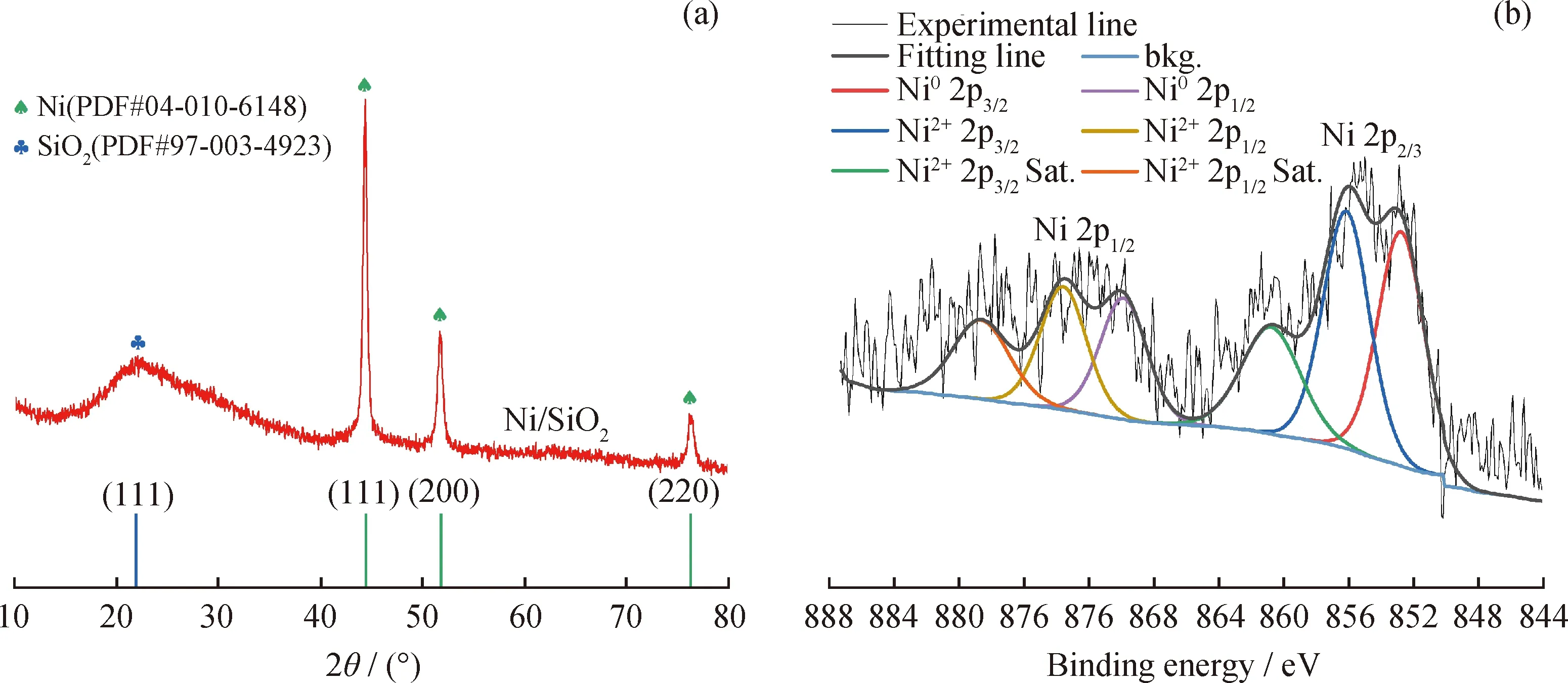
图1 Ni/SiO2催化剂的XRD谱图和Ni 2p的XPS图谱Fig.1 XRD patterns and Ni 2p XPS spectra of Ni/SiO2 catalyst(a)XRD;(b)XPS
由图1可知,XRD图谱在2θ为44.43°、51.78°、76.26°处出现的衍射峰分别对应于Ni的(111)、(200)、(220)晶面的特征峰。对于Ni 2p的XPS图谱,结合能在852.76和869.87 eV附近的特征峰分别归属于Ni02p3/2和Ni02p1/2,Ni0的质量分数为49.57%。催化剂的表征结果表明了催化剂表面存在还原态金属Ni物种。
1.1.2 实验方法
实验系统主要由介质阻挡放电(DBD)反应器、电源系统、测量系统等组成,示意图如图2所示。DBD反应器的内径为10 mm,外径为12 mm。用直径为3 mm的铜棒套在内径为3 mm、外径为4 mm的石英管,然后放置在反应器中心作为高压电极,反应器外壁包裹长为19 mm的铜片作为低压电极并接地。质量为1.5 g的Ni/SiO2催化剂包裹在石英棉内并填充在放电区域。CH4、CO2和He的流量通过质量流量控制器控制,掺混均匀后流入反应器。混合气(CH4、CO2和He体积比为15∶15∶70)的总流量为200 mL/min。实验系统的气压通过真空泵控制在16 kPa。反应温度在实验过程中通过管式炉控制并稳定在50~250 ℃(间隔50 ℃)范围内。纳秒脉冲电源(Xi′an Smart Maple Electronic Technology Co.,Ltd.,HVP-15P)的输出电压峰值为15 kV,最大放电频率和脉宽分别为100 kHz和1 ms。实验过程中使用高压探头(Rigol RP1018H,衰减比例1000∶1)和电流探头(Cybertek CP8030B,30 A/50 MHz)分别对施加在反应器上的电压和电流信号进行测量、采集。使用Agilent公司的7890B气相色谱仪对反应物和产物的组分和含量进行稳态测量,误差范围为±5%。

MFC—Mass flowmeter control;DBD—Dielectric barrier discharge;HV—High voltage side图2 实验系统图Fig.2 Schematic diagram of experimental setup
1.2 模拟部分
通过建立包含中性分子、自由基、激发态、带电粒子、吸附态等物质的详细动力学模型来模拟纳秒脉冲等离子体-催化剂协同CH4/CO2重整过程。基于零维模拟方法,将ZDPlasKin和CHEMKIN-II联偶,使得等离子体反应动力学和催化反应动力学解耦,实现对等离子体催化过程的求解。
等离子体催化放电过程中,由于丝状放电及表面电离波的存在[7],使得放电不均匀。因此,采用自洽的方法[7]计算约化电场强度E/N(V·cm2):
(1)
其中:E为电场强度(V/cm);N为中性粒子数密度(cm-3);β=P/V为功率密度(W/cm3);σ为电子电导率(S/cm);μe为电子的迁移率(cm2/(V·s));ne为电子的数密度(cm-3);e为电子的电荷量(1.602×10-19C)。
详细的等离子体催化CH4/CO2/He机理中的气相反应机理基于HP-Mech[8]进行构建。等离子体机理依据Sun等[9]的工作,更新和增加了CO2、CO、H2和H2O的电子碰撞反应、振动态物质参与的弛豫和链式反应以及O(1D)、O(1S)的相关反应路径。大多数的电子碰撞反应截面取自于LXCat数据库[10]。对于CO2的振动激发反应,其低能级的截面从Phelps数据库获取,高能级的截面基于第一非对称振动能级的截面通过Fridman approximation[11]计算得到。
基于Mohan等[12]提出的Ni基催化剂重整CH4/CO2机理,考虑等离子体中活性物质在催化剂表面的吸附、脱附以及表面反应,构建详细的等离子体催化反应机理。基于谐波过渡态理论,使用Eyring-Polanyi公式计算催化反应的速率常数k(cm2/s),如式(2)所示。
(2)
其中:kB为Boltzmann常数;T为催化反应温度(K),实验过程中反应温度通过管式炉定温控制,故不考虑等离子体放电造成的温升效应,催化反应温度与气相温度相同;h为Planck常数;ΔS#为活化熵(J/(mol·K));ΔH#为活化焓(J/mol);R为气体常数(8.3145 J/(mol·K))。
由于自由基较高的反应活性,在催化剂表面更容易直接吸附生成吸附态中间物质。直接吸附反应是在无活化焓垒的情形下发生,因此其速率常数kads(cm2/s)的计算公式简化如式(3)所示。
(3)
其中,σ为气相物质在催化剂表面的吸附系数。
CH4(v)、CO2(v)和H2(v)等振动态物质促进的解离吸附反应的速率常数kads,v(cm4/s)基于Fridman-Macheretα-model计算得到,如式(4)所示。

(4)
其中:α为振动态的能量利用效率,0<α<1;Ev是能级为v的振动态的能量(J/mol);H(x-x0)为Heaviside阶跃函数,当x>0时,H(x-x0)=1,当x<0时,H(x-x0)=0。
此外,催化反应机理考虑了气相物质与吸附态之间的E-R反应,自由基参与的E-R反应在吸附质与吸附态之间形成新的化学键之前没有发生化学键的断裂,因此自由基参与的E-R反应无需克服活化焓[13]。此时,E-R反应的速率常数(kER,cm2/s)可表达为如式(5)所示。
(5)
在本研究中,假设等离子体催化过程为均相反应体系,将表面吸附态物质当作气相物质进行处理。而基于以上公式计算得到的催化反应的速率常数的单位为cm2/s和cm4/s,通过乘以等离子体放电区域与催化剂颗粒表面积之比V/A转化为气相反应速率常数的单位cm3/s和cm6/s,计算得到的参数V/A约为0.018 cm。
2 结果与讨论
图3给出了不同放电电压和温度下,等离子体催化协同CH4/CO2重整过程中CH4和CO2消耗量的实验测量和模拟预测的对比。如图3所示,随着放电电压和温度的升高,约化电场强度增大,等离子体对CH4和CO2的离解和电离作用增强,促进反应物的消耗。
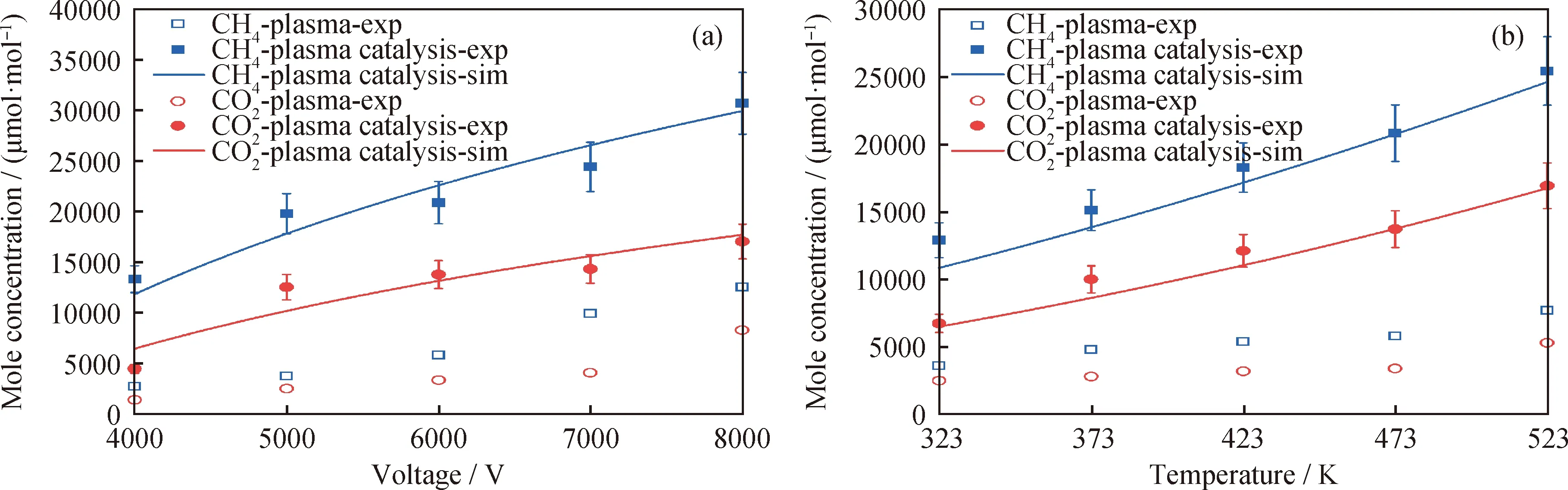
exp—Experiment;sim—Simulation图3 不同工况下CH4和CO2的实验测量和模拟预测的对比Fig.3 Comparison of CH4 and CO2 consumption between the experimental measurements and modeling prediction results at different conditions(a)Voltage;(b)Temperature Reaction conditions:(a)T=473 K;p=16 kPa;f=20 kHz;(b)p=16 kPa;U=6000 V;f=20 kHz
图4和图5为相应工况下生成物摩尔分数的实验测量和模拟结果的对比。由图4和图5可知,主要产物为合成气和C2H6,其中CO、H2的摩尔分数最高,在0.5%~2.8%之间,而C2H6的摩尔分数最高可达0.4%左右。实验中,C2H4、C2H2和C3H8等烃类的生成量较低,其摩尔分数在0.01%~0.10%之间。

exp—Experiment;sim—Simulation图4 不同工况下CO、H2和C2H6的实验测量和模拟结果的对比Fig.4 Comparison of species concentration for CO,H2 and C2H6 between experimental measurements and modeling results at different conditions(a)Voltage;(b)Temperature Reaction conditions:(a)T=473 K;p=16 kPa;f=20 kHz;(b)p=16 kPa;U=6000 V;f=20 kHz
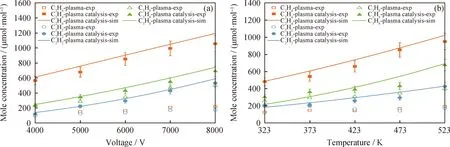
exp—Experiment;sim—Simulation图5 不同工况下C2H4、C2H2和C3H8的实验测量和模拟结果的对比Fig.5 Comparisons of species concentration for C2H4,C2H2 and C3H8 between experimental measurements and modeling results at different conditions(a)Voltage;(b)Temperature Reaction conditions:(a)T=473 K;p=16 kPa;f=20 kHz;(b)p=16 kPa;U=6000 V;f=20 kHz
从图3~图5可知,与单纯等离子体辅助CH4/CO2重整相比,通过等离子体与催化剂的协同,对CH4和CO2的活化转化能力大大增强,促进反应物的消耗和产物的生成。可以看到,稳态测量和数值模拟之间的误差在10%以内,验证了构建的动力学机理的合理性。由于本实验的产物在测量前并未通过冷凝装置收集液态产物,且气相色谱无法对含氧液态物质进行定性、定量分析,因此在本研究中并未给出液相含氧产物的对比结果。
图6(a)给出了E-R反应与相应的L-H反应的速率常数随温度变化。图6(a)中显示,振动态CO2(v)能够加快E-R反应CO2+H(s)→COOH(s)和CO2+H(s)→HCOO(s)的进程。在低温区间,E-R反应的速率常数明显高于L-H反应的速率常数。在473 K时,E-R反应CH3(s)+O→CH3O(s)的速率常数比相应的L-H反应CH3(s)+O(s)→CH3O(s)+Ni(s)的速率常数高6个数量级。因此,等离子体催化可以克服热催化甲烷干式重整反应热力学不利的影响,实现CH4和CO2的低温催化转化。
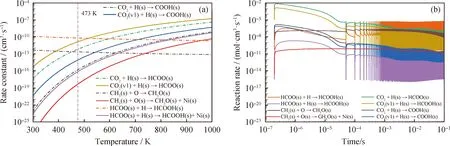
图6 E-R反应、L-H反应的速率常数随温度的变化以及速率随时间的变化Fig.6 Variation of rate constants for E-R and L-H reactions with temperature and time(a)Rate constant;(b)Reaction rate Reaction conditions:T=473 K;p=16 kPa;U=6000 V;f=20 kHz
图6(b)为温度473 K、压力16 kPa、峰值电压6000 V时E-R反应和相应的L-H反应的速率随时间的变化。图6(b)中显示CO2(v1)参与的E-R反应的速率与相应的CO2分子参与的E-R反应的速率具有相同的数量级,表明振动态CO2(v1)能显著加快E-R反应CO2(v1)+H(s)→COOH(s)/HCOO(s)的速率,促进COOH(s)和HCOO(s)的生成,进而通过L-H反应COOH(s)+Ni(s)→CO(s)+OH(s)和E-R反应HCOO(s)+H→HCOOH(s)促进吸附态CO(s)和HCOOH(s)的生成。同时,等离子体强化的E-R反应CH3(s)+O→CH3O(s)、HCOO(s)+H→HCOOH(s)的速率分别比相应的L-H反应CH3(s)+O(s)→CH3O(s)+Ni(s)、HCOO(s)+H(s)→HCOOH(s)+Ni(s)的速率高4和6个数量级,表明在等离子体与催化剂的协同作用下,自由基参与的E-R反应具有较高的反应活性,能有效改变并促进表面反应路径。
图7给出了温度473 K、压力16 kPa、峰值电压6000 V、放电频率20 kHz时的工况下等离子体催化协同甲烷干式重整过程中的表面反应路径。如图7所示,吸附态CH3(s)主要经CH4在催化剂表面的解离吸附生成,振动态CH4(v)通过解离吸附CH4(v)+2Ni(s)→CH3(s)+H(s)强化CH3(s)的生成,贡献率为1.6%。经E-R反应CH3(s)+H→CH4+Ni(s)重新生成CH4以及脱氢反应CH3(s)+Ni(s)→CH2(s)+H(s)是CH3(s)的主要消耗路径;其余的CH3(s)通过等离子体强化的E-R反应CH3(s)+O→CH3O(s)促进CH3O(s)的生成,对吸附态CH3O(s)的生成具有极强的促进作用。吸附态CH3O(s)通过加氢反应CH3O(s)+H→CH3OH(s)强化吸附态CH3OH(s)的形成,最后经脱附生成CH3OH。
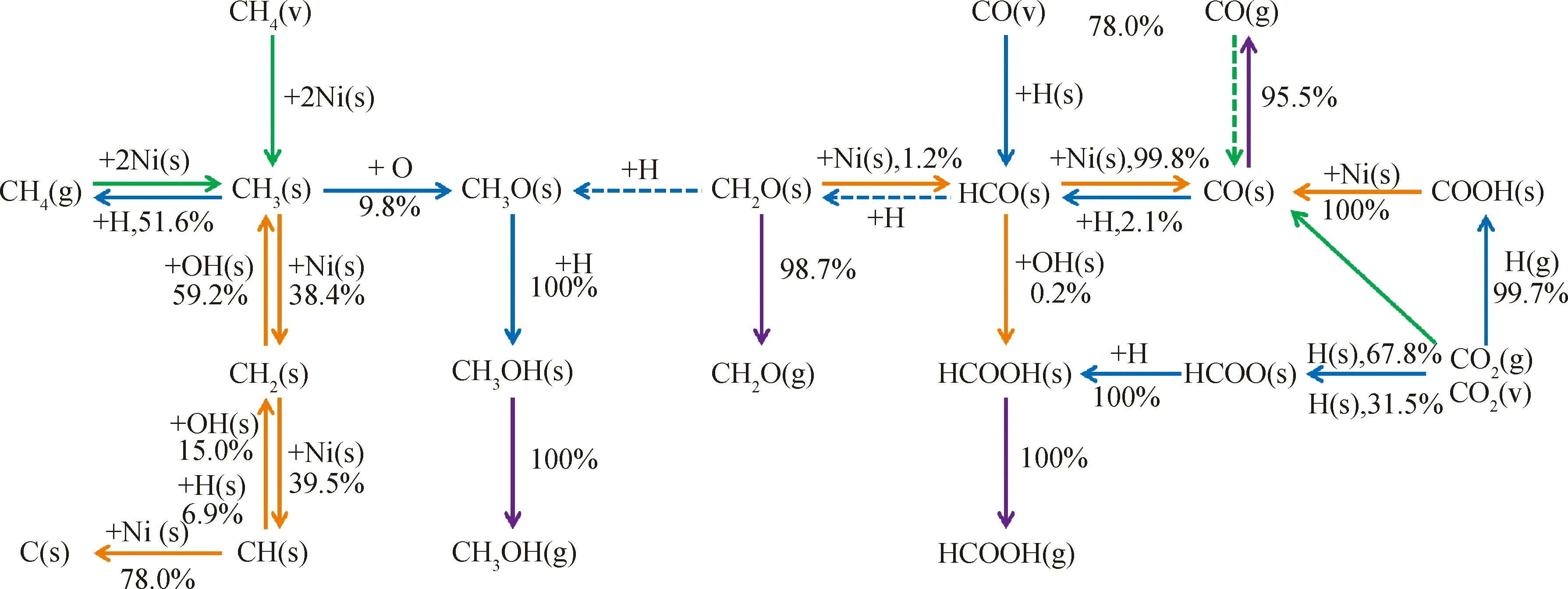
The scores in the figure are contribution rates of generation or consumption.图7 等离子体催化甲烷干式转化的表面反应路径Fig.7 Surface reaction path for plasma-catalytic dry reforming of methane Reactionconditions:T=473 K;p=16 kHz;U=6000 V;f=20 kHz
消耗路径显示,经L-H反应CH2(s)+OH(s)→CH3(s)+O(s)消耗的CH2(s)占其消耗总量的59.2%,而CH2(s)在催化剂表面脱氢生成CH(s)这一过程对CH2(s)的消耗的贡献率为39.5%。大量的CH(s)经氧化路径CH(s)+O(s)→C(s)+OH(s)脱氢生成积炭C(s),占其总消耗量的78.0%;部分CH(s)通过L-H反应CH(s)+OH(s)→CH2(s)+O(s)、CH(s)+H(s)→CH2(s)+Ni(s)加氢重新生成CH2(s)。因此,催化剂上的积炭主要通过CH4在活性位点的连续脱氢生成。
CO2参与的催化反应主要有2种类型,首先是在催化剂表面的解离吸附CO2+2Ni(s)→CO(s)+O(s);其次是E-R反应CO2+H(s)→COOH(s)和CO2+H(s)→HCOO(s)。振动态CO2(v)加快了E-R反应CO2(v)+H(s)→COOH(s)和CO2(v)+H(s)→HCOO(s)的反应速率,对COOH(s)和HCOO(s)的生成起到促进强化作用,分别占其总生成量的99.7%和31.5%。所有的COOH(s)在催化剂表面发生断键反应COOH(s)+Ni(s)→CO(s)+OH(s)生成CO(s)。大量的CO(s)经脱附反应生成CO,占其总消耗量的95.5%;部分CO(s)经E-R反应CO(s)+H→HCO(s)加氢生成HCO(s),绝大部分的HCO(s)通过振动态CO(v)与H(s)发生的E-R反应CO(v)+H(s)→HCO(s)生成,占其总生成量的97.2%。
吸附态HCO(s)主要在催化剂表面发生脱氢反应HCO(s)+Ni(s)→CO(s)+H(s)生成CO(s),占其总消耗量的99.8%,只有少量的HCO(s)通过与OH(s)发生L-H反应HCO(s)+OH(s)→HCOOH(s)+Ni(s)生成HCOOH(s),但其仅占HCOOH(s)总生成量的6.7%。图7显示,HCOOH(s)主要通过E-R反应HCOO(s)+H→HCOOH(s)生成,对HCOOH(s)的生成贡献率为93.3%,此路径同时是HCOO(s)的主要消耗路径,而生成的HCOOH(s)则进一步脱附生成HCOOH。
此外,HCO(s)与活性H原子发生E-R反应HCO(s)+H→CH2O(s)是吸附态CH2O(s)的主要生成路径,对CH2O(s)的生成贡献率为95.0%。绝大部分CH2O(s)在催化剂表面脱附生成CH2O,占其总消耗量的98.7%。
为了研究E-R反应的速率常数对等离子体催化CH4/CO2重整的产物浓度的影响,基于敏感性分析研究了活化焓(0.2、0.4、0.6 eV)对E-R反应速率常数进而对产物摩尔分数的影响。对数形式的敏感性系数S的定义如式(6)所示。
(6)
式中:S为敏感性系数;kj和k′j分别为不考虑和考虑活化焓时的E-R反应的速率常数;X和X′分别为不考虑和考虑E-R反应的活化焓时的产物摩尔分数。
图8为CO的敏感性分析。图8中显示反应CH3(s)+H→CH4+ Ni(s)对CO生成的促进作用最强。该反应是吸附态CH3(s)的主要消耗路径,通过此反应使得表面空白活性位点重生,进而通过HCO(s)的脱氢过程HCO(s)+Ni(s)→CO(s)+H(s)促进CO(s)的生成,最终经CO(s)的脱附生成CO。同时,反应HCOO(s)+H→HCOOH(s)和CH3O(s)+H→CH3OH(s)对CO的生成也呈现出较强的促进作用。其原因与上述反应CH3(s)+H→CH4+Ni(s)的促进机制类似,通过吸附态HCOOH(s)和CH3OH(s)的脱附重新生成空白活性位点Ni(s),以强化L-H反应HCO(s)+Ni(s)→CO(s)+H(s)的进程。
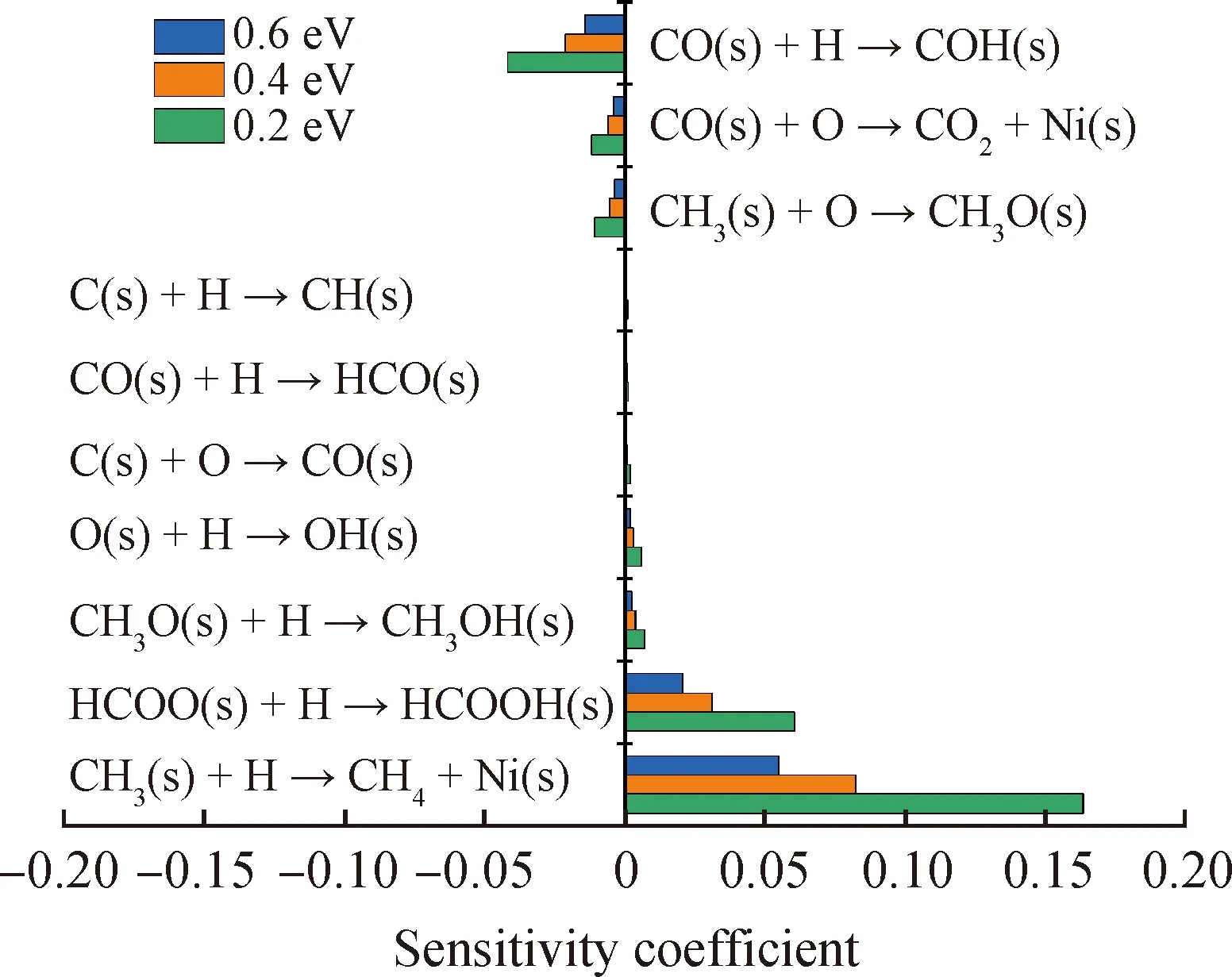
图8 CO的敏感性分析Fig.8 Sensitivity analysis of CO
反应CO(s)+H→COH(s)的负敏感性最大,这是因为此反应是吸附态CO(s)的次要消耗路径。同时,吸附态CO(s)的消耗过程CO(s)+O→CO2+Ni(s)对CO的生成也呈现出抑制作用。而反应CH3(s)+O→CH3O(s)之所以对CO的生成呈现出较强的负敏感性是因为该反应与E-R反应CH3(s)+H→CH4+Ni(s)对吸附态CH3(s)的消耗是竞争关系,通过抑制空白活性位点Ni(s)的重生而抑制吸附态CO(s)的生成。图8显示,E-R反应的活化焓越小,敏感性系数的绝对值越大,对CO的生成或消耗的影响越大。
3 结 论
采用实验测量和模拟预测相结合的思路,研究了NSD等离子体与Ni基催化剂协同CH4/CO2重整过程中的等离子体强化表面化学反应动力学。主要结论如下:
(1)通过等离子体与催化剂的协同,对CH4和CO2的活化转化能力远强于纯等离子体辅助CH4/CO2重整,促进反应物的消耗和产物的生成;等离子体催化协同CH4/CO2重整的主要产物为合成气和C2H6。
(2)等离子体强化的E-R反应CH3(s)+O→CH3O(s)、HCOO(s)+H→HCOOH(s)的速率分别比相应的L-H反应CH3(s)+O(s)→CH3O(s)+Ni(s)、HCOO(s)+H(s)→HCOOH(s)+Ni(s)的速率高4和6个数量级,表明在等离子体与催化剂的协同作用下,自由基参与的E-R反应具有较高的反应活性,能有效改变并促进表面反应路径。
(3)等离子体催化协同CH4/CO2重整过程中的表面反应路径主要以气相物质与吸附态物质之间的E-R反应为主,表明通过催化剂与等离子体的耦合,能够改变中间吸附态物质和产物的生成和消耗路径。敏感性系数的绝对值随活化焓的增大而减小,表明随着E-R反应速率常数的增大,其对物质生成或消耗的影响程度逐渐增强。
