婴儿早期发病的生物素-硫胺素反应性基底节病一例并文献复习
2023-09-19赵建闯易明胜黄先杰范亚珍崔晨航乔俊英
王 娜,赵建闯,易明胜,李 凡,黄先杰,范亚珍,崔晨航,乔俊英
1)郑州大学第三附属医院PICU科 郑州 450052 2)郑州大学第三附属医院放射科 郑州 450052
生物素-硫胺素反应性基底节病(biotin-thiamine responsive basal ganglia disease,BTBGD)也称硫胺素代谢功能障碍综合征2型(thiamine metabolism dysfunction syndrome 2,THMD2),是一种罕见的常染色体隐性遗传的神经代谢障碍性疾病,位于染色体2q36.3上的SLC19A3基因是其致病基因[1]。全球发病率不高,迄今为止,全球共报道100余例,国内报道更少。根据临床表现及发病年龄,BTBGD分为儿童起病的BTBGD、婴儿早期发病的Leigh样综合征或不典型的婴儿痉挛、成年起病的Wernicke′s样脑病,共3型[2]。现报道1例婴儿早期发病的BTBGD,分析其临床资料和测序结果,并进行文献复习,总结其临床特征,以期提高临床医师对该病的认识。
1 临床资料
1.1 一般资料患儿,男,1月龄,因“阵发性哭闹5天”收住于郑州大学第三附属医院PICU。5 d前患儿无明显诱因出现阵发性哭闹,初可安抚,后哭闹时间延长,不易安抚,伴吃奶量减少,哭闹严重时伴有双上肢抖动、角弓反张、双下肢硬性伸直、口唇青紫,不伴发热、咳喘、呕吐、腹泻;当地医院头颅CT(发病第4天)示颅内多发性对称性低密度灶,住院期间出现抽搐转至我院。患儿系第2胎第2产,胎龄39+3周,顺产,出生体重3 620 g,出生时无窒息史;母孕期体健,父母健康,非近亲结婚;胞哥6岁,发育正常;家族中无类似患者。
1.2 体格检查体重4 160 g,头围38 cm,身长51 cm,神志清,精神差,易激惹,面容正常,皮肤毛发无异常,前囟平软,双肺呼吸音对称,未闻及干湿性啰音。心音有力,律齐,心前区未闻及杂音,腹软,未触及包块,肠鸣音听诊正常,无注视,对声源无反应,颈软,四肢肌张力阵发性增高,双侧膝腱反射对称性引出,拥抱反射可引出,握持反射可引出,病理征阴性,脑膜刺激征阴性。
1.3 实验室检查肝肾功能、电解质、血同型半胱氨酸、血氨、甲状腺功能均未见异常。血串联质谱分析(MS/MS)及尿有机酸代谢筛查未见特异性异常,乳酸8.50~3.35 mmol/L(参考值0.70~2.20 mmol/L)。血气分析未见代谢性酸中毒。脑脊液外观淡黄色,清亮,细胞数正常;葡萄糖、氯正常,LDH 115.0 U/L(参考值0.0~40.0 U/L),蛋白1 102.0 mg/L(参考值120.0~600.0 mg/L),乳酸1.96 mmol/L(参考值1.00~2.90 mmol/L)。双侧眼底筛查未见异常。视觉诱发电位:双侧视P100未引出,脑干听觉诱发电位未见异常。动态脑电图:醒睡多量多灶性尖波、尖、慢波发放,额、颞及睡眠期显著。
1.4 影像学检查头颅彩超示脑实质回声弥漫性增强并脑室变窄(建议进一步排除脑水肿),双侧丘脑基底核回声异常。头颅MRI示双侧额顶枕叶、双侧丘脑、双侧基底节和双侧大脑脚、胼胝体压部、脑干、小脑蚓部多发基本对称性异常信号(图1)。
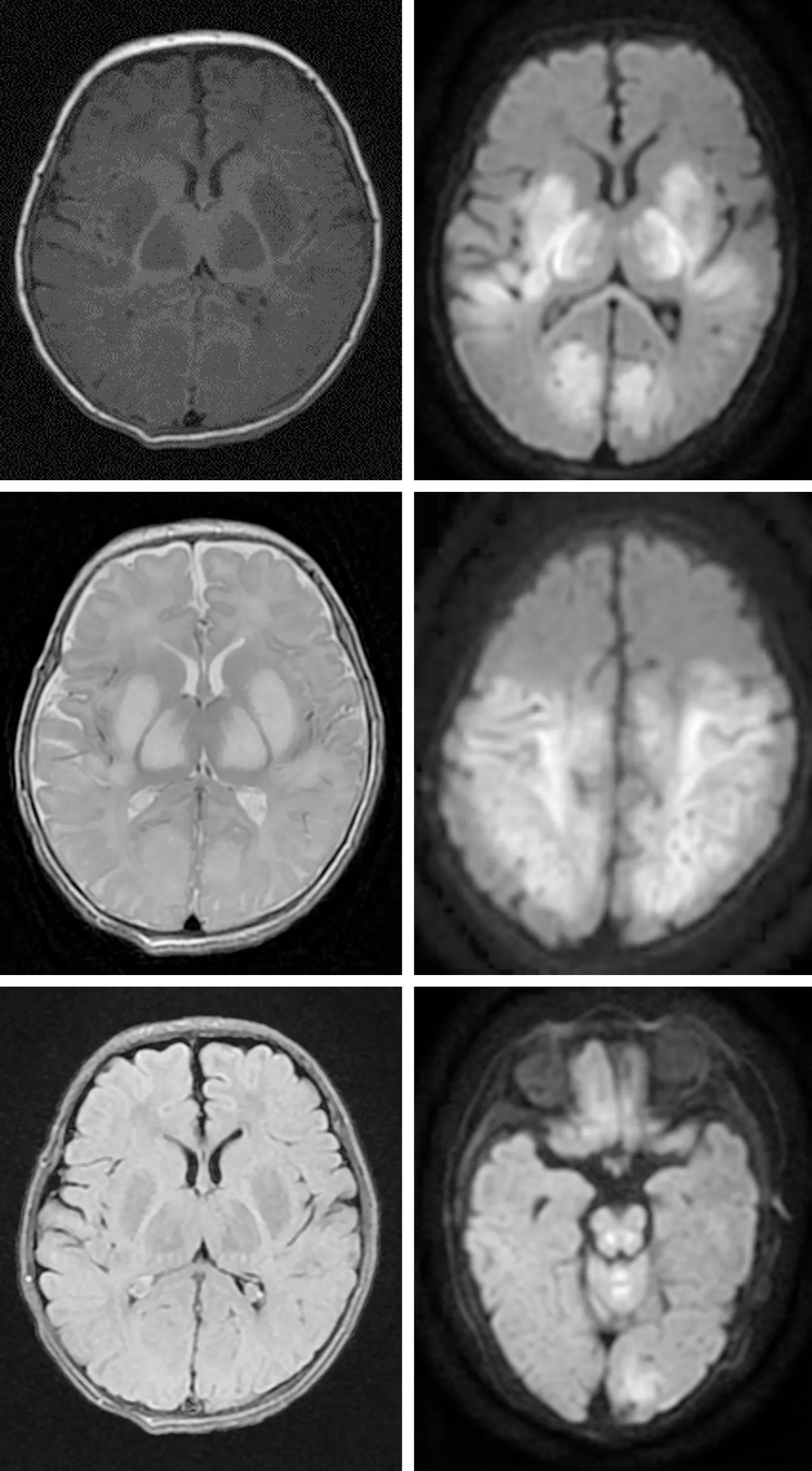
患儿1月龄,双侧基底节区、双侧丘脑及枕叶颞叶多发对称性T1WI低信号(左上)、T2WI高信号(左中),T2Flair呈稍低信号(左下),DWI序列(右上、右中、左下)可见双侧额顶枕颞叶、双侧基底节区、双侧丘脑及脑干、小脑蚓部及胼胝体压部呈明显高信号
1.5 基因测序经患儿父母签署知情同意书,并经医院伦理委员会批准(批准文号2022-178-01),行基因检测,由北京迈基诺医学检验所完成。结果(图2)显示,患儿存在SLC19A3全基因杂合缺失和c.842G>A(p.W281X)半合子突变,为复合杂合突变。前者来自母亲,已有报道;后者来自父亲,为首次发现。
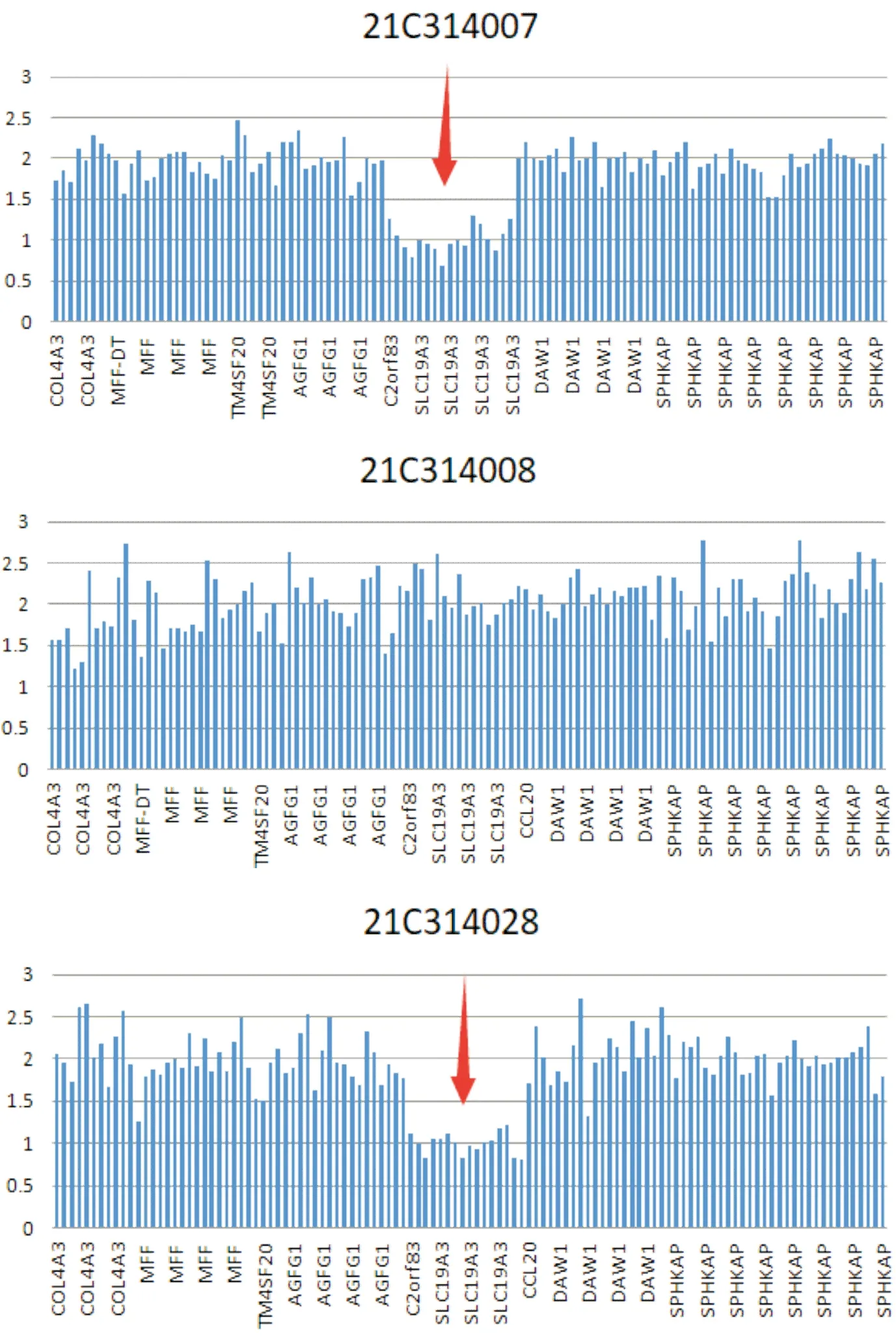
患儿(21C314007):SLC19A3基因全基因杂合缺失和c.842G>A(p.W281X)半合子突变;母(21C314028):全基因杂合缺失;父(21C314008):c.842G>A(p.W281X)半合子突变
1.6 治疗及随访入院后给予鸡尾酒疗法(辅酶Q10、左卡尼汀、多种维生素)及生物素6 mg/(kg·d)、硫胺素25 mg/(kg·d)及托吡酯治疗。治疗第3天患儿易激惹状态好转,哭闹减少,吃奶及睡眠逐渐恢复正常,睡眠逐渐增多,未见抽搐发作,乳酸较入院时下降,但仍偏高,好转出院。院外规律服用上述药物。1个月后(2月10天),复查动态脑电图示醒睡各期各导未见弥漫性或局限性慢波活动,未见癫痫样波,正常范围脑电图。视觉诱发电位示双侧视觉通路功能重度异常。头颅MRI示脑萎缩,弥漫性脑软化并角质增生,双侧额颞顶部硬膜下积液,双侧视神经T2WI信号增高(图3)。继续给予生物素、硫胺素、辅酶Q10、左卡尼汀及托吡酯治疗。6月龄复诊,体格发育正常,头围小,颅缝重叠,发育落后,注视、追视差,追听有反应,不会抬头、竖头不稳,不会翻身,不会坐,不会抓物,四肢肌张力高,吃奶好,睡眠好,间断出现点头、拥抱状,有时伴有尖叫,成串发作,每次10次左右,复查头颅MRI示双侧额部扁平,双侧额颞顶部硬膜下积液,双侧顶部硬膜下出血,弥漫性脑软化并胶质增生,双侧视神经纤细(图3)。动态脑电图提示不典型高峰失律,加用左乙拉西坦联合上述药物治疗,痉挛发作较前减少。
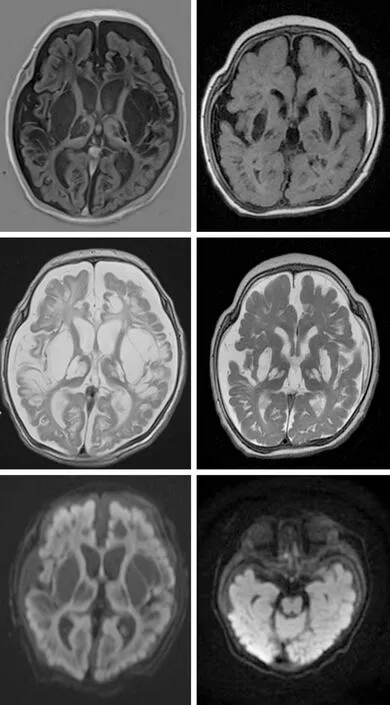
患儿2月10天,双侧基底节区、双侧丘脑及额颞枕叶多发脑软化灶,病灶呈T1WI低信号(左上),T2WI高信号(左中),DWI序列未见明显弥散受限(左下)。6月龄时(右上、右中、右下),双侧基底节区、双侧丘脑脑软灶范围较前明显缩小,部分病灶周围见胶质增生,脑干及小脑蚓部未见明显异常信号,提示局部好转
1.7 文献复习以“生物素-硫胺素反应性基底节病”“生物素反应性基底节病”“硫胺素代谢综合征紊乱综合征2型”为检索词检索万方数据知识服务平台、CNKI。以“biotin responsive basal ganglia disease”“biotin-thiamine responsive basal ganglia disease”“thiamine transporter-2 deficiency”或“SLC19A3”为检索词检索PubMed。为避免遗漏,在确定为BTBGD相关文献后再手动查询婴儿早期发病的文章。检索起止时间均为建库至2022年3月31日。排除病史不详、资料不足及重复病例后,共15篇文献[3-17]累计43例(包括本例患儿)婴儿早期发病的BTBGD病例纳入分析。
43例来自31个家庭,男28例,女15例。26例(60.5%)有父母近亲婚配史或家族中多人患病或不良孕产史。在地域分布上,亚洲占53.4%(23/43),非洲占25.6%(11/43),欧洲16.3%(7/43),北美洲4.7%(2/43)。亚洲病例中,47.8%在中国(11/23)。
43例中,33例(76.7%)在生后3个月内发病,22例(51.2%)出现易激惹、哭闹过度,31例(72.1%)伴有肌张力异常,30例(69.8%)伴有喂养困难或吞咽困难,29例(67.4%)伴有癫痫发作,11例(25.6%)伴有嗜睡、反应减少,100%(43/43)存在发育落后。
43例(100%)头颅MRI示双侧基底节可见对称性异常信号。包括本病例有8例行视神经检查,5例视神经萎缩,1例视神经正常,2例视觉诱发电位提示双侧视P100未引出。
34例行血乳酸检测,其中24例同时行脑脊液乳酸检测。79.4%(27/34)血乳酸升高,33.3%(8/24)脑脊液乳酸升高。27例血乳酸升高患儿中18例死亡,7例血乳酸正常患儿中4例死亡。
27例(62.8%)携带SLC19A3基因纯合变异,16例(37.2%)携带复合杂合变异。
19例给予生物素联合硫胺素治疗,6例死亡,8例加重,5例好转。4例仅给予生物素治疗,3例死亡,1例加重。3例仅给予硫胺素治疗,均死亡。17例未给予治疗,16例死亡,1例加重。
2 讨论
婴儿早期发病的BTBGD多在生后3个月内发病,大多伴有肌张力异常、喂养困难或吞咽困难、癫痫发作,易激惹、哭闹过度,也有嗜睡、反应减少等表现,100%存在发育落后。SLC19A3基因突变的表型差异很大,即使是同一家系相同基因型的患者,表型也可以明显不同[4,18]。乳酸酸中毒很常见,但脑脊液乳酸酸中毒与血乳酸升高无相关性。有研究[9,19]表明乳酸酸中毒往往表明预后不佳。文献复习中18/27例血乳酸升高患者死亡,4/7例血乳酸正常患者死亡,但死亡与是否干预、干预的时机、干预药物的种类及剂量等多种因素相关,需要进一步多中心研究分析。
婴儿早期发病的BTBGD头颅MRI双侧基底节可见对称性异常信号。Kevelam等[10]根据核磁共振模式,把BTBGD疾病过程分成4个阶段,包括急性期、急性后期、中间期和终末期。急性期大脑皮质和白质、小脑白质、胼胝体、基底核、丘脑、脑干和小脑白质等多个区域均出现严重肿胀和T2高信号,DWI弥散受限。急性后期大脑白质肿胀部分消退,伴有稀疏和(或)囊性变性,主要在皮质下白质、基底核和丘脑。中间期脑干、丘脑、基底核、大脑白质和胼胝体出现不同程度的萎缩及囊肿样结构,并伴有大脑皮质的广泛变薄,之前DWI弥散的区域不受限,部分可见硬膜下血肿。终末期表现为大脑白质和皮质、胼胝体、丘脑、基底核、小脑、中脑等弥漫性萎缩。这四个阶段彼此迅速跟进,大脑异常在几周到几个月中演变。本例患儿头颅MRI的变化相与上述一致:早期表现为双侧额顶枕叶、双侧丘脑、双侧基底节和双侧大脑脚、胼胝体压部、脑干、小脑蚓部可见多发片状长T1长T2信号,DWI高b值弥散受限呈高信号;后期头颅MRI提示脑萎缩、弥漫性脑软化并胶质增生,双侧额颞顶部硬膜下积液,双侧顶部硬膜下出血。不同时期的头颅MRI检查可以为疗效评估及预后判断提供参考。
文献复习结果显示,阿拉伯人群中最常见的变异是SLC19A3基因5号外显子纯合错义突变c.1264A>G(p.T422A),临床表现为经典型的BTBGD[14]。而我国以复合杂合变异为主,且为散发。本例患儿携带有SLC19A3基因整体杂合缺失和c.842G>A(p.W281X),后者为首次报道,丰富了SLC19A3的基因变异数据库。
对于BTBGD患儿的治疗尚无统一方案。在早期的治疗经验中,单独给予大剂量生物素治疗后可以逆转临床症状,因此BTBGD也被称为生物素反应性基底节病[20]。考虑到硫胺素、生物素均为水溶性维生素,治疗安全性较好,因此对于临床疑诊的BTBGD患儿应尽早开始治疗,不必等待基因测序结果。疾病急性期可酌情增加药物剂量,患者需终身服药,在病情稳定后,硫胺素及生物素可采用小剂量替代治疗。
综上所述,对于婴儿期不明原因出现异常哭闹,有肌张力增高或抽搐的脑病伴有基底节对称性病变的患儿,尤其对于3个月龄以内的患儿,除了考虑线粒体脑病,还应考虑BTBGD。对于高度怀疑BTBGD者,可早期给予生物素、硫胺素治疗,同时进行相关基因检测以明确诊断,改善患者预后。
