基于结构深度网络嵌入方法的微生物-疾病关联关系预测
2023-09-18陈亚丽雷秀娟
陈亚丽, 雷秀娟
(陕西师范大学 计算机科学学院,陕西 西安 710119)
与人体有关的微生物包含真核生物、古生菌、细菌、真菌和病毒[1],它们主要寄生在人体的皮肤、生殖器、口腔,特别是肠道[2]等部位。研究发现,微生物在人体中的数量大约为细胞数量的10倍[3],这表明人体内微生物群落相对庞大。
许多研究表明,微生物群落与复杂疾病有关。例如,肠道内细菌可通过对肠黏膜上皮细胞作用,如影响DNA完整性、调节免疫反应、诱导炎症反应等,促进结直肠癌发生[4-6]。孤独症患者的肠道菌群和正常人有着显著的区别,其中白色念珠菌和一些促进炎症发生的梭菌水平明显升高。而且,胃肠道感染破伤风梭菌会增加孤独症的患病风险,这种梭菌会释放神经毒素,而毒素可能会通过迷走神经传递到大脑,从而引起孤独症的症状[7-9]。对于帕金森患者而言,其肠道内的肠杆菌科越高,症状往往就越严重[10]。因此,确定微生物与疾病之间的关系已成为当前生物信息学领域的一个重要研究课题。
考虑到微生物与人类疾病的紧密关系。国内外研究者已经启动了许多有关微生物和疾病研究的项目并且建立了相关数据库。为了能够系统地了解微生物组成及其在疾病中的重要性,美国和欧盟联合发起了一项人类微生物组计划(human microbiome project,HMP)[11]。此外,还建立了人类微生物-疾病关联数据库(human microbe-disease association database,HMDAD),该数据库目前包括39种疾病、292种微生物以及483种微生物与疾病的关联[12]。通过生物医学实验验证来确定微生物与疾病之间的关系是一项费力且昂贵的工作。因此,基于HMDAD数据库,人们提出了多种计算模型用于预测微生物与疾病的关联关系。这些模型大致可以分为三类:第一类是基于评分函数的计算模型。例如,Chen等[12]基于KATZ度量提出了预测微生物-疾病关系的第一个模型KATZHMDA,该模型根据微生物-疾病网络中两个结点间的行走步长和行走次数来计算这两个结点的关联概率。Li等[13]提出了一种基于双向加权网络的计算模型BWNMHMDA来预测微生物-疾病关系,与KATZHMDA不同的是,该计算模型在计算结点间的相似性时考虑了路径的起点和终点。Bao等[14]提出了一种基于网络一致性投影的计算模型NCPHMDA来预测微生物-疾病关联。Huang等[15]提出了一种基于深度优先搜索算法的计算模型PBHMDA来预测与疾病有关的潜在微生物。虽然上述方法都获得了比较好的预测效果,但绝大多数与疾病相关的微生物还是未知的。因此,Fan等[16]通过整合多源数据和基于路径的HeteSim评分,建立了一个预测微生物-疾病关联的新模型MDPH_HMDA。第二类是基于网络算法的模型。例如,Shen等[17]提出了一种异构网络上的随机游走算法RWRH来预测微生物和疾病的关联。Luo等[18]提出了一种基于随机游走的计算模型NTSHMDA,与RWRH方法不同的是,该计算模型结合了网络拓扑相似性,将异构网络上微生物与疾病的关联分配不同的权重。Niu等[19]提出了一种在超图上进行随机游走的计算模型RWHMDA,与RWRH方法不同的是,RWHMDA保留了疾病和微生物之间多对多的关系。Yan等[20]提出了一种基于相似性和双随机游走的预测模型BRWMDA来预测潜在的微生物和疾病关联。第三类是基于机器学习的计算模型。例如,Peng等[21]提出一种基于自适应增强的计算模型ABHMDA来预测微生物与疾病的关联关系,该模型利用由具有相应权重的弱分类器组成的强分类器来计算微生物与疾病的关联概率。Wang等[22]基于拉普拉斯正则化最小二乘分类器构建一个半监督计算模型LRLSHMDA来预测微生物与疾病的关联。Liu等[23]基于图正则化非负矩阵分解提出一个新的计算模型NMFMDA来预测微生物与疾病的关联。尽管这些计算模型能够取得比较稳定的预测性能,但是它们所依赖的已知微生物和疾病关联数据都是基于HMDAD数据库的,该数据库规模较小,所以在预测微生物和疾病的关系时具有一定的局限性。
本文提出一种基于网络嵌入的计算模型(NEMDA)来预测微生物和疾病的关联关系。首先,通过已知的微生物-疾病关联构建一个二分网络,用一种网络嵌入方法-结构深度网络嵌入在该二分网络上学习结点嵌入。其次,计算微生物功能相似性、微生物相互作用谱相似性和疾病语义相似性、基于症状的疾病相似性,分别来表示微生物和疾病的生物学特征,并结合微生物和疾病嵌入特征得到新的特征来表示微生物-疾病对。最后,用深度神经网络构建预测模型,并将该特征作为深度神经网络的输入来计算微生物与疾病的关联概率。实施交叉验证和案例分析来评估NEMDA模型的预测能力。实验结果表明,在不同的参数影响下,NEMDA模型的结果比较稳定,预测性能高于其他6种比较方法。
1 材料和方法
本文提出NEMDA模型来预测微生物-疾病的关联,图1为NEMDA模型的总体框架图,该模型主要由3个步骤构成:首先,整合HMDAD和Disbiome数据库,将得到的已知微生物-疾病关联构建一个二分网络;然后,在该二分网络上使用结构深度网络嵌入(structural deep network embedding,SDNE)提取结点特征,结合微生物功能相似性、微生物相互作用谱相似性以及疾病语义相似性、基于症状的疾病相似性得到微生物-疾病对的特征;最后,使用得到的微生物-疾病对的特征训练深度神经网络(deep neural network,DNN)模型,来预测新的微生物-疾病关联。
1.1 材料
1.1.1 人类微生物-疾病关联
本文使用的数据来自2个数据库:一个是人类微生物-疾病关系数据库[24](HMDAD,http://www.cuilab.cn/hmdad),该数据库包含39种疾病、292种微生物以及483个经实验验证微生物-疾病关联。另一个是Disbiome数据库[25](https://disbiome.ugent.be/),该数据库不断更新并收录微生物-疾病关系数据,截至2020年12月,该数据库从已知文献以及不同的数据库中共收集了350种疾病和1 581种微生物以及8 695个经实验验证的微生物-疾病关联。本文对这2个数据库进行整合,以扩大已知的微生物-疾病关联。首先,根据不同的证据以及检测方法,一个微生物-疾病对可能会被多次记录,因此需要过滤掉这些重复的记录。其次,由于一些疾病会存在多个名称,但是它们拥有相同的概念唯一标识符(concept unique identifier, CUI),比如Liver cirrhosis和Cirrhosis是肝硬化的两种不同形式,但它们的CUI号均为C4228437。因此,在整合数据时,需要对其进行标准化以避免重复。综上,本文首先在NCI Metathesaurus(NCIm) 数据网站对所有的疾病搜索对应的CUI号;其次,对所有疾病-微生物关联根据疾病的CUI号去冗余;最后,共得到254种疾病、1 519种微生物以及7 258个微生物-疾病关联。本文进一步构建微生物-疾病关联邻接矩阵B∈Rnm×nd,其中,nm表示微生物的数量,nd表示疾病的数量,如果微生物结点m(i)和疾病结点d(j)有已知的关联,则Bij=1,否则Bij=0。
1.1.2 疾病语义相似性
依据文献[27]的方法,本文计算两种疾病的语义相似性。具体来讲,基于MeSH描述符,每种疾病可以通过有向无环图(directed acyclic graph,DAG,记作GDA)表示。对于疾病d而言,其有向无环图可描述为GDA=(d,Td,Ed),其中Td表示包括d在内的所有祖先结点集合,Ed表示相应的边集合。在GDA中,疾病a对于疾病d的语义贡献值定义为
(1)
式中:Δ是语义贡献因子,用来表示Ed集合中连接疾病a和疾病a′的边的权重。通常情况下,在GDAd中,距离d越远的疾病,它的语义贡献值越小。在这里,Δ设置为0.5,a、a′∈Td。疾病d的语义计算公式为
VD(d)=∑a∈TdDd(a)。
(2)
如果在两个疾病的DAG中,祖先结点的交集越多,则表明这两种疾病越相似,基于这样的假设,疾病di和dj的语义相似性可定义为
(3)
式中:Ddi(a)和Ddj(a)分别表示a对疾病di和dj的语义贡献;VD(di)和VD(dj)分别表示疾病di和dj的语义值。
1.1.3 基于症状的疾病相似性
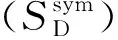
1.1.4 微生物功能相似性
本文使用misim[27]方法来计算微生物的功能相似性。假设dk为一个特定的疾病,那么DK={dk1,dk2,…,dkm}是表示与微生物相关的一组疾病。根据1.1.2,疾病dk与DK的语义相似性可定义为
(4)
式中m是指与微生物相关的疾病个数。

(5)
1.1.5 微生物相互作用谱相似性
基于相似微生物更倾向于与相似的疾病有关的假设[28],本文进一步构建了微生物的Jaccard相似性。对于一个具体的微生物mi,它的相互作用谱向量定义为P(mi),该向量表示微生物mi与所有疾病的相互作用关系,如果某一疾病与微生物mi有已知关联,则P(mi)相对应位置的值为1,否则为0。
Jaccard相似性是一种常用的统计方法,可用于计算两个样本之间的相似性。因此,这里采用Jaccard相似性来表示微生物mi和mj的相互作用谱相似性,其可定义为
(6)

1.1.6 疾病的生物学特征表示
为了更加充分地利用疾病语义相似性和基于症状的疾病相似性,本文通过整合这两种相似性来表示最终的疾病相似性,则对于疾病di和dj来说,其相似性定义为
(7)

1.1.7 微生物的生物学特征表示
与疾病类似,通过整合微生物的功能相似性和相互作用谱相似性来表示微生物的最终相似性,则微生物mi和mj的相似性定义为
(8)

1.1.8 微生物-疾病对的特征表示
在微生物-疾病关系预测问题上,多数研究依赖于微生物和疾病的生物学特征,如微生物功能相似性、疾病语义相似性和基于症状的疾病相似性,很少有研究考虑在微生物-疾病二分网络上提取特征。考虑到在二分图上提取特征有利于保持网络性质,本文从已知的微生物-疾病关联网络中学习特征。因此,本文使用3种类型的特征向量表示微生物-疾病对,一种是基于微生物相似性的特征向量,一种是基于疾病相似性的特征向量,还有一种是基于结构深度网络嵌入学习得到的特征向量。
1.2 方法
1.2.1 基于SDNE网络嵌入的特征提取
近年来,图嵌入[29-31]即网络表征学习成为复杂网络分析方面的研究重点,其目的是用一个低维、稠密的向量去表示图中的点,并且该向量能够反映网络的结构。本质上来说,网络中的两个结点共享的邻近结点越多,即这两个结点的上下文越相似,两个结点对应的低维向量距离越近。图嵌入的最大好处是可以将学习到的向量输入到任何机器学习模型去解决具体问题。基于此,越来越多的人将图嵌入方法应用到各个研究领域,目前应用比较广的领域有推荐、链路预测、结点分类等。常用的图嵌入方法有DeepWalk[32]、LINE[33]、Node2vec[34]、SDNE[35]等。与其他嵌入方法相比,SDNE不进行随机游走,在实践中比较稳定。因此,本文使用SDNE在已知微生物-疾病网络中学习结点的特征。

一阶相似性是指成对结点之间的相似性或者结点与其邻近结点的相似性。本文用邻接矩阵G中的某一行Gi={Gi1,Gi2,…,Gi(nm+nd)}表示结点i与其他结点之间的相似度。如果Gij>0,表示结点i、j之间存在正的一阶相似性,否则结点i、j之间的一阶相似性为0。二阶相似性是指一对结点的邻域结构的接近程度,它刻画的是Gi、Gj之间的相似性。SDNE在微生物-疾病关联网络提取特征的框架如图2所示。

图2 SDNE提取特征示意图Fig.2 Schematic diagram of SDNE feature extraction注:网络版为彩图。
将结点向量Gi作为模型的输入,通过自编码器对其进行降维压缩,表示为Zi,然后再重建特征,其损失函数定义为
(9)
值得注意的是,邻接矩阵G是一个稀疏阵,其中零元素的个数远远多于非零元素的个数,如果直接将G作为自动编码器的输入,则更容易重构G中的零元素。为了解决这个问题,文献[33]使用带权损失函数,对于非零元素具有更高的惩罚系数。修改后的损失函数为
(10)
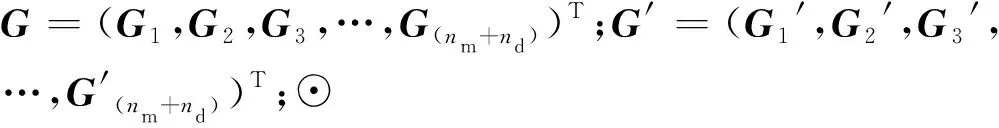
与LINE类似,SDNE也想保持一阶和二阶相似性,并且将两者同时优化,以便同时捕获局部成对相似性和结点邻域结构相似性。对于一阶相似性的计算,首先利用深层自编码器得到隐层向量Z,其次计算左侧嵌入Zi和右侧嵌入Zj之间的距离。因此,损失函数定义为
(11)
式中Zi和Zj分别表示结点i、j的低维嵌入向量。
SDNE为了保证一阶相似性和二阶相似性,提出了半监督学习的方法,结合上面的监督学习和无监督学习,联合后的损失函数为
(12)
式中:α为控制一阶损失的参数;γ为控制正则化项的参数;Lreg是一个L2范数正则化项,用来防止过拟合,其计算公式为
(13)
式中:K表示隐层的个数;W(k)和W′(k)分别表示编码器和解码器第k层的权重向量。
将SDNE方法应用到微生物-疾病二分网络上,可以得到一个(nm+nd)×d的嵌入矩阵MD,其中d表示结点嵌入的维数,MD的每一行MDi,i=1,2,…,(nm+nd)表示相应的nm个微生物结点和nd个疾病结点的嵌入表示。
1.2.2 关系预测
DNN模型由三部分组成:一个输入层、多个隐藏层、一个输出层[36-38]。输入层接收DNN的输入,通过多个隐藏层以非线性方式进行转化,最后输出到输出层。每个隐藏层都应该根据前一层的输出提取更多概括性的特征,因此DNN在分类问题上表现出优越的区分能力[39-41]。本文将微生物-疾病预测问题转化为二分类问题,将从SDNE输出的特征与微生物和疾病的生物学特征结合得到(2d+nm+nd)维的特征向量,并将这些特征输入到DNN中,以训练模型。训练结束后,如果给定微生物-疾病对的预测概率超过阈值,则说明存在相应的微生物-疾病关联。
对于DNN网络结构的设计,本文使用一种常见的塔模型结构,其中隐藏层第一层的神经元最多,下一层的神经元少于上一层。当前层h的每一个神经元连接上一层h-1的所有神经元,每个隐藏层的输出可以用以下公式计算:
(14)
式中:xi、xi+1分别表示第i、i+1个隐藏层的输出;wi、bi分别表示第i个隐藏层对应的权重和偏置。在输入层和隐藏层,使用ReLU激活函数。在输出层,使用Sigmoid激活函数,得到每一个标签对应的概率值。这里使用均方误差作为损失函数,Adadelta优化算法用于最小化损失函数。为避免过拟合,将Dropout用于输入层和隐藏层,根据经验,本文将Dropout取值为0.5。
2 结果分析
本文将通过交叉验证和案例分析来验证NEMDA模型的预测性能,并与4种分类器以及6种预测方法进行对比。此外,还对NEMDA模型中的一些参数进行分析。
2.1 评价指标
受试者工作特征(receiver operating characteristic,ROC)曲线反映的是真实工作率(true positive rate,TPR,记作RTP)和错误工作率(false positive rate,FPR,记作RFP)之间的关系,该曲线的横坐标表示错误工作率,纵坐标表示真实工作率。RTP和RFP分别依据公式(12)和(13)进行计算:
(15)
(16)
式中:TP和TN分别表示预测正确的正样本和负样本;FP和FN分别表示预测错误的正样本和负样本。通常用ROC曲线下的面积(area under ROC curve,AUC,记作AUC)来评估模型的预测性能,AUC值越高,模型的性能越好。
PR曲线表示的是精准率(precision,记作Pr)和召回率(recall,记作Rc)之间的关系,它的横坐标表示召回率,纵坐标表示精准率。PR曲线下的面积(area under PR curve,AUPR,记作AUPR)可以用来评估模型的预测性能,AUPR值越大,表明模型的预测性能越好。通过设置不同的阈值可以计算Pr和Rc的值,计算公式如下:
(17)
(18)
这里,参数TP、FP和FN的定义与上面一致。
2.2 参数分析
一些重要的参数会影响模型的性能,本文主要针对从SDNE中提取的特征维数,以及DNN层数进行参数敏感性分析。设置SDNE特征维数依次为32、64、96、128、160;设置DNN隐藏层神经元的个数依次为1 024、512、256、128、64等,隐藏层层数依次为2、3、4、5、6。本文采用五折交叉验证并且综合考虑SDNE特征维数和隐藏层层数对于NEMDA模型性能的影响,具体做法如下:采用网格搜索穷举遍历所有的参数组合,并计算每组参数值对应的AUC值。
从图3可以看出,随着特征维数的增加,NEMDA模型的性能会提高,因为维数增加可以编码更多有用的信息。但是,当维数超过128,再增加特征维数时,NEMDA模型的AUC值逐渐减小,预测性能开始缓慢下降。产生这一结果的原因可能是特征维数过大,导致在编码时引入了一些噪声。此外,隐藏层层数在一定程度上也影响NEMDA模型的性能;随着隐藏层层数增加,模型的性能大多呈下降趋势。当SDNE嵌入特征维数设置为128、隐藏层层数设置为3时,NEMDA模型的性能最好。因此,在实验中将特征的维数设置为128,隐藏层层数设置为3。
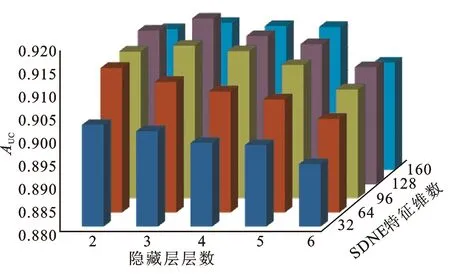
图3 综合考虑SDNE嵌入维数和隐藏层层数对于NEMDA预测性能的五折交叉验证Fig.3 Cross validation of NEMDA prediction performance considering SDNE embedding dimension and number of hidden layers comprehensively注:网络版为彩图。
2.3 不同类型特征的比较
在NEMDA中,本文结合SDNE得到的嵌入特征和微生物与疾病的相似性特征来表示微生物-疾病对的特征,然后使用DNN进行预测。为了验证本文使用的特征表示信息的有效性,这里分别将结合后的特征、SDNE得到的特征以及相似性特征作为微生物-疾病对的特征,讨论它们各自对NEMDA模型性能的影响。在五折交叉验证下,实验得到的ROC曲线和PR曲线如图4、5所示。 将结合后的特征(红线表示)与仅使用相似性特征(蓝线表示)的结果进行对比,说明SDNE得到的嵌入特征能够提高模型的预测性能;将结合后的特征(红线表示)与仅使用SDNE得到的特征(绿线表示)的结果进行对比,说明结点的相似性特征对于提高模型的预测性能是非常重要的。综上,结合SDNE得到的嵌入特征和微生物与疾病的相似性特征来共同表示微生物-疾病对的特征时,NEMDA模型可以获得更好的预测性能。
图4 五折交叉验证下不同类型特征比较的ROC曲线Fig.4 ROC curves for comparing different types of features under cross validation注:网络版为彩图。
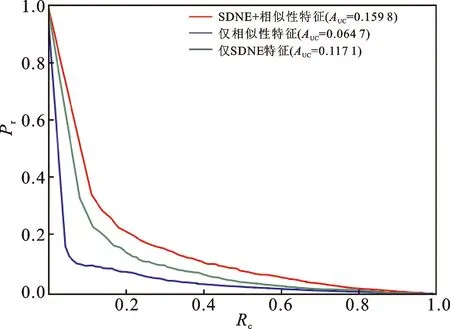
图5 五折交叉验证下不同类型特征比较的PR曲线Fig.5 PR curves for comparing different types of features under cross validation注:网络版为彩图。
2.4 与其他分类器比较
本文使用DNN模型作为分类器来预测微生物-疾病的关联关系,为了评估该模型的性能,将DNN模型与一些常用的分类器算法进行比较,包括线性回归(Linear regression)、逻辑回归(Logistic regression)、自适应增强(AdaBoost)和梯度下降树(gradient boosting decision tree,GBDT)。
通过五折交叉验证来评估各个分类器的预测性能,结果如图6、7所示。使用DNN模型(AUC=0.916 8,AUPR=0.159 8)作为分类器的预测效果比使用线性回归(AUC=0.882 5,AUPR=0.061 1)、逻辑回归(AUC=0.855 6,AUPR=0.117 8)、自适应增强(AUC=0.819 6,AUPR=0.107 5)、梯度下降树(AUC=0.903 7,AUPR=0.156 7)好,结果表明DNN模型适合解决微生物-疾病预测问题。
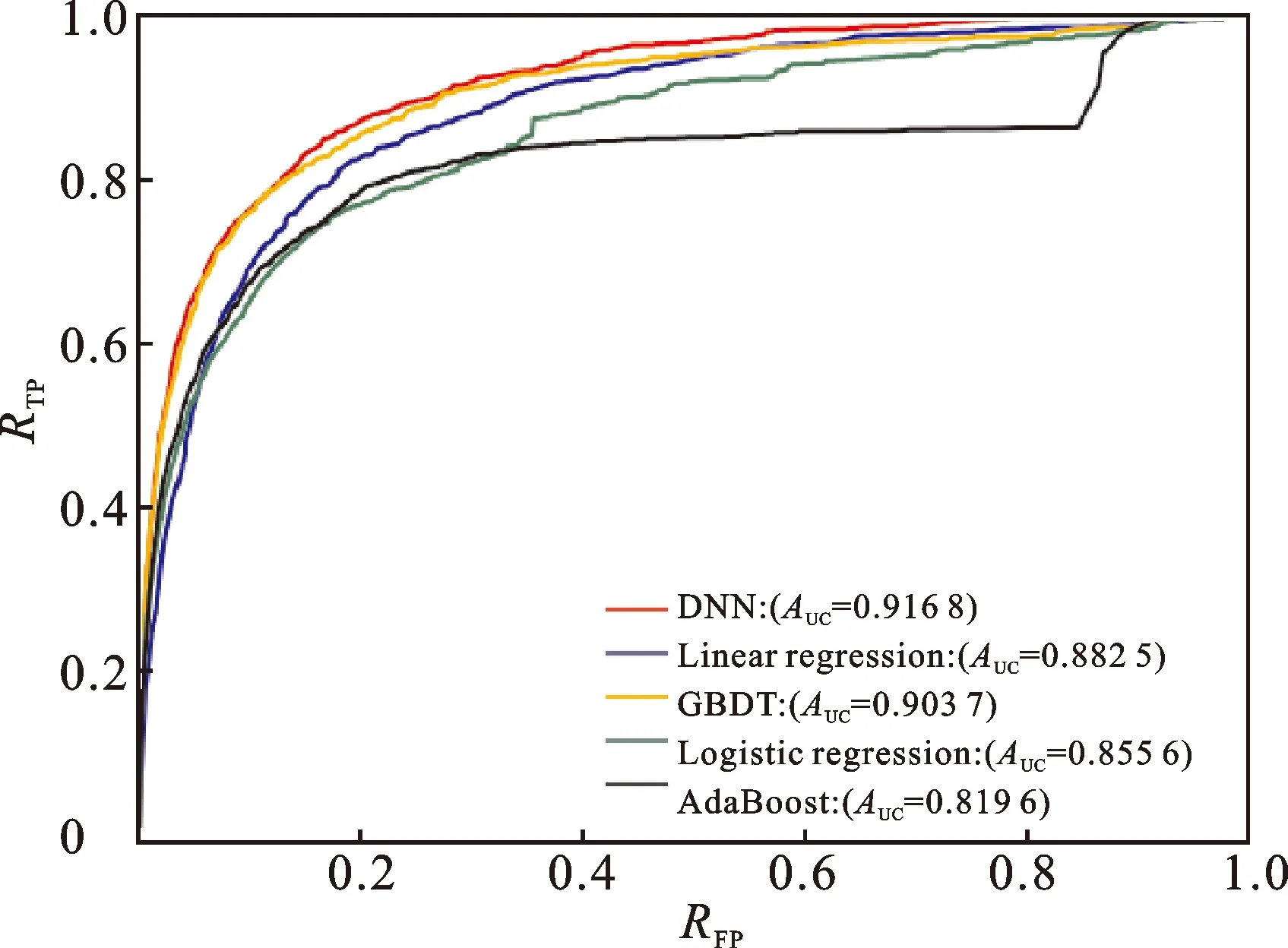
图6 五折交叉验证下不同分类器性能比较的ROC曲线Fig.6 ROC curves for comparing the performance ofdifferent classifiers under cross validation注:网络版为彩图。

图7 五折交叉验证下不同分类器性能比较的PR曲线Fig.7 PR curves for comparing the performance ofdifferent classifiers under cross validation注:网络版为彩图。
2.5 与其他方法比较
为了评估NEMDA模型的预测性能,本文将其与一些经典的微生物-疾病预测方法进行对比,包括KATZHMDA[12]、NCPHMDA[14]、LRLSMDA[22]、PBHMDA[15]、NTSHMDA[18]和BRWMDA[20]。通过五折交叉验证来评估NEMDA模型与其他6种比较方法的预测性能,比较结果如图8、图9所示。6种比较方法的AUC值分别是KATZHMDA(0.672 9)、NCPHMDA(0.733 7)、LRLSHMDA(0.866 4)、PBHMDA(0.878 4)、NTSHMDA(0.675 8)、BRWMDA(0.819 1),本文方法的AUC值为0.916 8,高于6种比较方法的AUC值。6种比较方法的AUPR值分别是KATZHMDA(0.056 3)、NCPHMDA(0.104 2)、LRLSHMDA(0.144 4)、PBHMDA(0.165 9)、NTSHMDA(0.068 7)、BRWMDA(0.072 1),本文方法的AUPR值为0.159 8,高于6种比较方法的AUPR值。由于本文所用的数据集是对HMDAD数据库和Disbiome数据库的整合,即将之前HMDAD数据库中的39种疾病、292种微生物以及450个已知的关联关系扩充到254种疾病、1 516种微生物以及7 190个已知的微生物-疾病关联,因此相比之前的邻接矩阵,本文的邻接矩阵变得非常大且稀疏,所以KATZHMDA、 NCPHMDA和NTSHMDA方法的效果并不是很理想;LRLSHMDA方法作为一种机器学习的方法,本身就适用于大数据集,所以预测效果比较好;而PBHMDA方法是对微生物-疾病网络进行深度优先搜索,从而捕获更加有用的信息,所以PBHMDA方法的预测效果也比较好。实验结果表明,本文提出的NEMDA模型在微生物-疾病预测问题上表现良好,是一种有效的预测工具。

图8 五折交叉验证下不同方法的AUC值和ROC曲线Fig.8 AUC values and ROC curves of different methods under cross validation注:网络版为彩图。

图9 五折交叉验证下不同方法的AUPR值和PR曲线Fig.9 AUPR values and PR curves of different methods under cross validation注:网络版为彩图。
2.6 案例分析
为了进一步评估NEMDA方法在识别新的微生物-疾病关联的预测能力,本文分别对哮喘、炎症性肠病(inflammatory bowel disease,IBD)和结直肠癌(colorectal cancer,CRC)这3种疾病进行案例分析。对于每一种疾病,首先删除与该疾病具有已知关联的微生物,然后根据NEMDA对候选微生物的预测得分进行降序排序,最后验证前20种微生物与所研究疾病之间的关联是否被相关文献证实。
哮喘是一种以慢性气道炎症为特征的异质性疾病[42]。全球大约有3亿多人患有哮喘,而且在1990—2015年期间,哮喘的发病率增加了12.6%[43]。随着16rRNA测序技术的发展,人们已经发现哮喘和微生物组之间有着重要的关系。在本文中,当采用NEMDA预测潜在的微生物-疾病关联时,前20名候选微生物中有12个被PubMed中的相关文献验证(如表1所示)。例如,在哮喘患者和健康对照组的痰液对比中发现,在哮喘患者肠道内,Lachnospiraceae(第1)[44]、Actinobacteria(第17)[45]过度表达,Sutterella相对丰度降低。此外,有实验证明梭状芽胞杆菌,包括Ruminococcus、Blautia(第3)、Clostridium和Subdoligranulum(第20)的类群丰富度和丰度与哮喘呈正相关(P<0.05)[46]。采用16rRNA和定量PCR(qPCR)技术分析3个月大儿童的肠道菌群组成,与健康对照组相比,在哮喘儿童肠道内Clostridium(第5)数量增加[47],Faecalibacteriumprausnitzii(第13)水平降低[48]。有实验表明,Shigella(第7)在患有固定气道阻塞的哮喘患者的肠道内富集[49],哮喘的严重程度与粪便中嗜黏液Akkermansia(第15)水平成负相关[50],在中性粒哮喘病患者中,Porphyromonas(第14)相对丰度降低[51]。

表1 与哮喘有关的排名前20的微生物Tab.1 Top 20 microorganisms related to asthma
IBD是一种特发性肠道炎症性疾病,主要包括溃疡性结肠炎(ulcerative colitis, UC)和克罗恩病(Crohn disease, CD),临床表现为腹痛、腹泻和血便等,该病很难根治,容易反复发作,有潜在的癌变风险。因此,本文对IBD进行了案例分析,以评估NEMDA对于新的微生物-疾病关联的预测能力。结果如表2所示,在前20个候选微生物中有19个得到相关文献验证。例如,在IBD患者体内Bifidobacterium、Lactobacillus(第1)[52]和Acinetobacter(第19)[53]数量增加,但是Roseburia(第4)[54]和Lachnospiraceae(第3)[55]相对丰度降低。此外,在IBD患者的唾液微生物群中观察到,Streptococcus(第2)、Prevotella、Neisseria(第18)、Haemophilus、Veillonella(第9)和Gemella在很大程度上导致了菌群失调[56]。在患有艰难梭菌感染(clostridium difficile infection,CDI)的IBD患者中,Dorea(第8)水平降低[57]。有实验证明,与健康对照组相比,在CD患者体内,Bacteroides、Eubacterium(第20)、Faecalibacterium和Ruminococcus明显减少,但是Fusobacterium(第11)[58]、Halomonas、Shewanella和Streptococcus、Actinomyces(第10)[59]明显增多。在IBD患者粪便中,Shigella(第12)增加,而Akkermansia(第17)[60]、Firmicutes和Collinsella(第16)[61]减少。

表2 与炎症性肠病有关的排名前20的微生物Tab.2 Top 20 microorganisms related to inflammatory bowel disease
CRC是一种常见的恶性肿瘤,每年会导致近70万人死亡,成为全球第四大最常见的致命癌症[62]。虽然癌症很大程度上是由于遗传和环境因素引起的,但越来越多的证据表明,与人体相关的微生物菌群的失调在形成炎症环境和促进肿瘤生长和扩散方面发挥着作用[63]。同样地,当采用NEMDA预测新的微生物-疾病关联时,前20个候选微生物中有17个得到相关文献验证(如表3所示)。例如,有研究表明,与CRC相关的基因有LCN2和DUOX2,其基因表达与结直肠癌相关细菌的丰度有关,比如Ruminococcacea和Veillonella(第1)[64]。此外,Bilophila、Parabacteroides和Odoribacter(第8)与早期发病的CRC有关[65]。在CRC患者的肠道菌群中,Enterococcus、Escherichia/Shigella(第2)、Klebsiella、Streptococcus和Peptostreptococcaceae(第15)相对丰度增加[66]。在浸润性CRC患者中,Corynebacterium(第14)、Enterococcus、Neisseria、Porphyromonas和Sclegelella的相对丰度较高[67]。宏基因组分析表明,CRC患者肠道内的Proteobacteria(第5)和Firmicutes(第16)发生了显著的改变[68],无高脂血症或高胆固醇血症的CRC患者与Bilophila(第13)和Butyricicoccus(第19)这两种细菌密切相关[69]。研究者进一步验证CRC与微生物代谢产物之间的关系,通过实验发现,在CRC患者的肠腔内,与代谢紊乱和宿主代谢有关的微生物菌群增加,如Prevotellaceae和Coriobacteriaceae(第11)[70],并且CRC与微生物代谢物产物具有较强的相关性,主要以Proteobacteria(第5)和Actinobacteria(第18)为主[71]。

表3 与结直肠癌有关的排名前20的微生物Tab.3 Top 20 microorganisms associated with colorectal cancer
3 结语
微生物对于人类健康与疾病起着关键性的作用。微生物-疾病关联不仅可以揭示疾病的发病机理,而且可以促进疾病的诊断和预后,因此对于微生物-疾病关联的研究受到了广泛关注。本文提出一种新的计算模型NEMDA来预测潜在的微生物-疾病关联,五折交叉验证和案例分析结果均表明NEMDA能有效地预测微生物-疾病关联。
NEMDA获得较好的预测性能主要是依赖以下几个方面:第一,整合HMDAM和Disbiome数据库,构建了一个更大的微生物-疾病关联网络,使得NEMDA计算模型能够充分利用已知的关联;第二,使用SDNE在微生物-疾病二分网络上提取特征,充分利用微生物-疾病网络的性质;第三,计算微生物的功能相似性、微生物的相互作用谱相似性和疾病的语义相似性、基于症状的疾病相似性分别表示微生物和疾病的生物学特征,并且结合SDNE从微生物-疾病网络中提取的特征表示微生物-疾病对的特征,然后采用DNN模型进行预测,判断微生物-疾病对是否有关联。
当然,NEMDA仍然存在一些缺陷,需要在未来的工作中解决。比如,本文只用到微生物-疾病组学数据,在未来的工作中还可以整合其他的组学数据,比如疾病与药物组学数据、微生物与药物组学数据等,来提高NEMDA模型的预测性能。由于SDNE运行速度较慢,本文只进行了五折交叉验证,在未来的工作中可以通过改进算法的复杂度来解决此问题。
