Integrating microRNAs and mRNAs reveals the hormones synthesis and signal transduction of maize under different N rates
2023-09-16YUEKaiLILinglingXIEJunhongZechariahEFFAHSumeraANWARWANGLinlinMENGHaofengLILinzhi
YUE Kai ,LI Ling-ling #,XIE Jun-hong ,Zechariah EFFAH,Sumera ANWAR ,WANG Lin-lin ,MENG Hao-feng ,LI Lin-zhi
1 College of Agronomy,Gansu Agricultural University,Lanzhou 730070,P.R.China
2 State Key Laboratory of Aridland Crop Science,Gansu Agricultural University,Lanzhou 730070,P.R.China
3 Council for Scientific and Industrial Research,Plant Genetic Resources Research Institute,Bunso 00233,Ghana
4 Department of Biosciences,Durham University,Durham DH1 3LE,UK
5 Department of Botany,Government College,Women University,Faisalabad 38000,Pakistan
6 College of Resources and Environment Science,Gansu Agricultural University,Lanzhou 730070,P.R.China
Abstract
The effect of nitrogen (N) fertilizer on the development of maize kernels has yet to be fully explored.MicroRNA-mRNA analyses could help advance our understanding of how kernels respond to N.This study analyzed the morphological,physiological,and transcriptomic changes in maize kernels under different N rates (0,100,200,and 300 kg ha–1).The result showed that increasing N application significantly increased maize grains’ fresh and dry weight until N reached 200 kg ha–1.Higher levels of indole-3-acetic acid,cytokinin,gibberellin,and a lower level of ethylene were associated with increased N applications.We obtained 31 differentially expressed genes (DEGs) in hormone synthesis and transduction,and 9 DEGs were regulated by 14 differentially expressed microRNAs (DEMIs) in 26 pairs.The candidate DEGs and DEMIs provide valuable insight for manipulating grain filling under different N rates.
Keywords: maize kernels,phytohormones,high-throughput sequencing,microRNA
1.Introduction
Nitrogen (N) is an important regulator of maize kernels; however,insufficient or excessive N application is not conducive to grain development.Insufficient N leads to poor soil,while excessive N destroys the soil’s ecological environment (Bot and José 2005),which is a prevalent problem worldwide.Generally speaking,maize crop uses only 30–50% of the applied N fertilizer,while most of the N is left in the soil as nitrate and ammonia N or in the atmosphere as nitrous oxide (Hodgeetal.2000; Kuanetal.2016).Unsuitable N applications have brought great challenges to crop production and limited the sustainable development of agriculture.
Proper grain development is important for the formation of a high yield.However,grain development is a complex process,and the synthesis of starch and protein substances is the most important process affected by various hormone signals (Bernardietal.2019; Panigrahietal.2019; Wang and Zhang 2020).Auxin (IAA),abscisic acid (ABA),cytokinin (CTK),ethylene (ETH),gibberellin (GA),brassinosteroids (BRs),jasmonic acid (JA),and salicylic acid (SA) are the main hormones involved in grain development (Zhangetal.2016; Tengetal.2021).
Understanding the expression of hormone-related genes involved in grain development is the first step in exploring the molecular regulation of hormones in grain formation.Previous researchers showed that IAA is the main factor regulating crop grain development.Indeed,some IAA-regulated genes and regulatory signals of grain size have also been identified,such asAuxinResponseFactor6(ARF6),AuxinResponseFactor8(ARF8),BigGrain1(BG1),andSmallOrganSize1(SMOS1) (Ayaetal.2014; Liuetal.2015; Naetal.2019).Nonhebeletal.(2020) found high contents of IAA and ABA in grains are conducive to grain filling,and some specific genes controlling IAA and ABA synthesis were also explored,including9-cis-epoxycarotenoiddioxygenase2(NCED2),YUCCA9(OsYUC9),andYUCCA11(OsYUC11).Inhibited expression of theZmACO2gene and reduced ETH content improved maize yield (Ningetal.2021).In addition,the high content of ETH can inhibit the synthesis of amino acids in rice grains (Xuetal.2022).
Advances in bioinformatics have helped identify small RNAs and explore the relationships between small RNAs and genes.Despite their small size,small RNAs can regulate grain development by regulating target genes.For example,Houetal.(2020) found that ata-miR172c-3p,osa-miR171a,miR396s,and tae-miR160 take part in grain filling by regulating hormones.In maize,miR167 and miR528 families may be involved in the metabolism process and adaptation to the environment during grain filling (Lietal.2016).Recently,microRNA and mRNA integration analysis has been applied to crop grain development (Lietal.2013; Zhangetal.2019; Houetal.2020; Yangetal.2021).The construction of microRNA and mRNA co-expression networks is particularly important for understanding the molecular mechanism of maize kernel development.
Hormones are very important for grain filling.However,the physiological and molecular mechanisms by which N regulates the synthesis and signaling of hormones are still unclear.Identifying N-sensitive genes will help understand the molecular mechanism of hormone regulation of grain development under different N conditions and genetically improve the spring maize through gene transformation,obtaining high-yield maize that can thrive with reduced N inputs.Therefore,this study analyzed the changes in the grain transcriptome under different N application rates and determined the physiological indicators of hormone changes.This study provides valuable information for the functional study of N efficient regulation of maize varieties.It also presents abundant candidate microRNAs and mRNAs for the innovation of maize N utilization germplasm resources and the cultivation of excellent varieties.
2.Materials and methods
2.1.Site description,plant materials,and physiological measurement
The study was conducted at the Dingxi Experimental Station (35°28´N,104°44´E) of Gansu Agricultural University,Gansu Province,Northwest China.The experimental site was located in the semi-arid Loess Plateau,with an average altitude of 2 000 m above sea level and an annual frost-free period of 140 days.The aeolian soil is classified as a Calcaric Cambisol.At a depth of 0–20 cm,it possesses a sandy loam texture with 50% sand,moderately low fertility,slightly alkaline pH (8.3),soil organic carbon of 7.65 g kg–1,and Olsen’s phosphorus of 13 mg kg–1.Annual cumulative air temperature >10°C is 2 240°C,and annual radiation is 5 930 MJ m–2,with 2 480 h of sunshine per year.The average annual growing season precipitation at the site is 443.2 mm.
Four N fertilization levels,N0 (no N fertilizer),N1 (100 kg N ha–1),N2 (200 kg N ha–1),and N3 (300 kg N ha–1),were applied using a completely randomized block design with three replications per treatment.One-third of the total N fertilizer (urea,46% N) was applied at sowing,while the remaining two-thirds were at the six-leaf collar stage (V6).Phosphorus (150 kg ha–1) was applied as P2O5.No potassium fertilizer was applied as available potassium (220 mg kg–1) in the soil is enough for maize production.The area of each plot was 18.7 m2(4.4 m×4.25 m).Completely film mulched alternate narrow and wide ridges with furrow planting were used in this experiment.The alternate wide (0.7 m) and narrow (0.4 m) ridges were mulched with transparent plastic films (1.1 m wide) before sowing maize.The seeds of maize cultivar Xianyu 335,which is the most popular variety in this area,were sown in furrows at a target final stand of 52 500 plants ha–1in late April and harvested in early October of each year.
After 21 days of pollination (DAP),three ears were labeled from each plot,and 200 grains were then randomly sampled from the middle part of the three ears,counting from the 10th to the 20th row of ears (bottom to top),and divided into two portions.The first portion of grains was frozen in liquid N at once,carried back to the laboratory,and stored in a –80°C fridge for RNA and hormones analysis.From the second portion of grains,the weight of 100 grains was taken.Then,the grains were dried in an oven for 30 min at 105 and 80°C to constant weight and reweighed.
The endogenous concentration of ABA,IAA,GA,CTK,and ETH was determined by enzyme-linked immunosorbent assay (ELISA).The ELISA kits were supplied by Jiangsu Meibiao Biotechnology Co.,Ltd.(Yancheng,China).
2.2.mRNA and small RNA analysis
The frozen kernels from N0,N1,and N2 treatments were used for sequencing and analysis of small RNA and mRNA (N3 treatment was skipped based on non-significant differences in kernels weight between N2 and N3)
RNA extractionTotal RNA was extracted using Trizol (Life Technologies,USA),according to the manufacturer’s instructions.The purity of total RNA was detected by NanoDrop (NanoDrop 2000,Life Technologies,USA),and the integrity of RNA was detected by Agilent 2100 (Agilent Technologies,USA).
Small RNA sequencing and analysisSmall RNAsequencing (RNA-seq) of the nine libraries as conducted on Illumina HiSeq 2500 sequencing platform at Gene Denovo Biotechnology Co.,Ltd.(Guangzhou,China).After removing low-quality reads,all of the clean tags were aligned with small RNAs in the GenBank database (Release 209.0) and Rfam database (11.0) and with the reference genome.Then,the clean tags were searched against the miRBase database (Release 21) to identify known microRNAs.Novel microRNAs were predicted by the Software Mireap_v0.2 according to their genome positions and hairpin structures.
mRNA sequencing and analysisRNA-sequencing (RNA-seq) of the nine libraries was conducted on Illumina HiSeq 2500 sequencing platform at Gene Denovo Biotechnology Co.,Ltd.(Guangzhou,China).Briefly,raw reads were filtered,and the clean reads were mapped to the maize reference genome (B73 RefGen_v4).
Construction of DEMI-target (DEM) networkBased on a false discovery rate (FDR)<0.01 and |log2FC|>1,we selected the DEGs.Similarly,we identified microRNAs with |log2FC|≥2 andP-value<0.05 in comparison as significant DEMIs.The relationships between DEGs and DEMIs were predicted by the Software PatMatch (v1.2).
qRT-PCR analysisTotal RNA was extracted from cells using TRIzol reagent (Sigma,USA) according to the manufacturer’s protocol and then stored at –80°C until further use.The cDNA was prepared using the RevertAidTMFirst Strand cDNA Synthesis Kit (Thermo Fisher Scientific Inc.,USA) following the manufacturer’s protocol.After that,qRT-PCR was performed to identify the expression patterns of selected genes in the two groups.To amplify fragments corresponding to the selected genes,we designed primers using the NCBI Primer-BLAST tool.We used Zm00001d041948 as an internal control to normalize the expression level of the target genes.In preparation for the qRT-PCR,we prepared a 20-μL reaction volume containing 10 μL of the Real Master Mix SYBR,8 μL of ddH2O,1 μL each of the forward and reverse primers,and 1 μL of cDNA.
2.3.Statistical analysis
The data for all dependent variables were analyzed by analysis of variance using SPSS 25.0.Treatments were compared using Duncan’s multiple range test,and the significance level was set toP<0.05.Graphs were plotted in Origin 2019 and Cytoscape 3.9.1.
3.Results
3.1.Physiological characteristics of maize grain under different N rates
With the increase in N application,the grain dry weight and fresh weight increased,but no significant difference was observed between N2 and N3 (Fig.1).N1 and N2 significantly increased grain dry weight by 23.6 and 92.3% and increased grain fresh weight by 27.2 and 52.0%,respectively,compared with N0.The IAA,ABA,CTK,and GA contents increased with the N application; however,the ETH content decreased (Fig.2).There was no significant difference in ABA,CTK,and ETH contents at N1,N2,and N3,but N increased the ABA and CTK contents while decreasing the ETH content as compared to N0.N2 and N3 significantly increased IAA and GA contents compared with N0 and N1.N2 increased IAA,ABA,CTK,and GA contents by 29.7,8.1,18.8,and 14.3%,respectively,and decreased ETH by 17.3%,compared to N0.In addition,the dry weight of 100 kernels showed a significantly positive correlation with IAA,ABA,CTK,and GA and was negatively correlated with ETH content in grains.
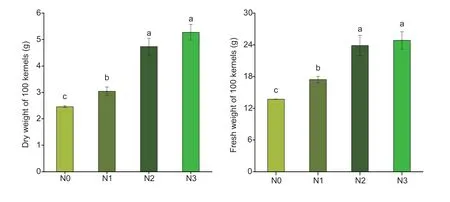
Fig.1 The morphology of maize kernels under different N rates.N0,no N fertilizer; N1,N2,and N3 indicate 100,200,and 300 kg N ha-1,respectively.Bars mean SD (n=3).Different letters indicate significant differences at P<0.05.
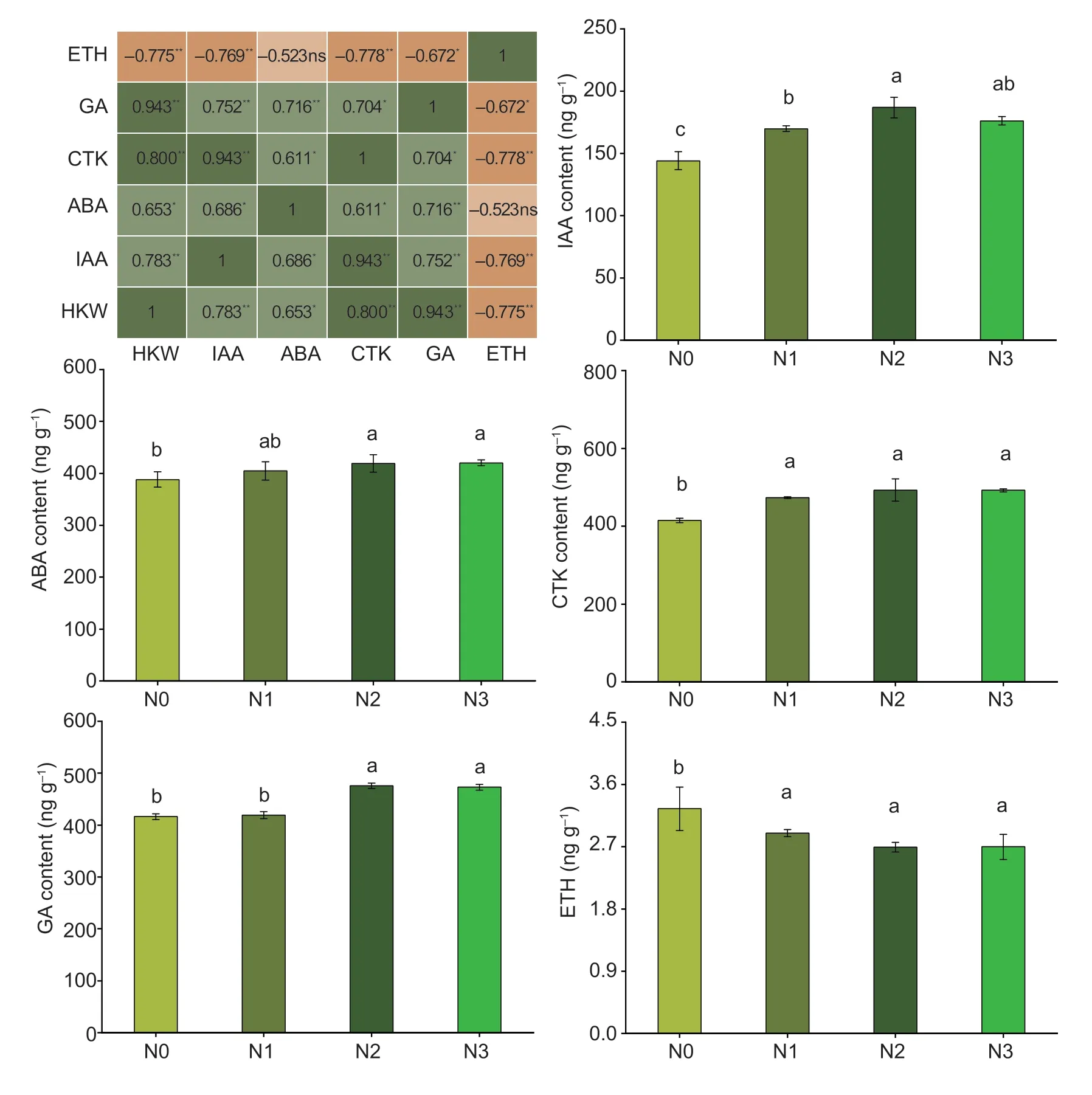
Fig.2 Correlation between the dry weight of 100 kernels and hormones,and hormone contents of maize kernels under different N rates.N0,no N fertilizer; N1,N2,and N3 indicate 100,200,and 300 kg N ha-1,respectively.HKW,dry weight of 100 kernels; IAA,indole-3-acetic acid; ABA,abscisic acid; CTK,cytokinin; GA,gibberellin; ETH,ethylene.Bars mean SD (n=3).Different letters indicate significant differences at P<0.05.ns,not significant at P<0.05; *,significant at P<0.05; **,significant at P<0.01.
3.2.Analysis of differentially expressed microRNAs (DEMls) and differentially expressed genes (DEGs)
To understand the functions of the microRNAs under different N rates in maize grains,sequence data were gathered from 9 samples.Clean tags were obtained after eliminating reads containing reads without 3´ adapter,reads with 5´ adapter,reads without inserts,insert reads with fragment lengths less than 18 nt,and Poly A.On average,13.8,14.4,and 13.8 million clean tags were obtained in N0,N1,and N2 treatments,respectively (Table 1).
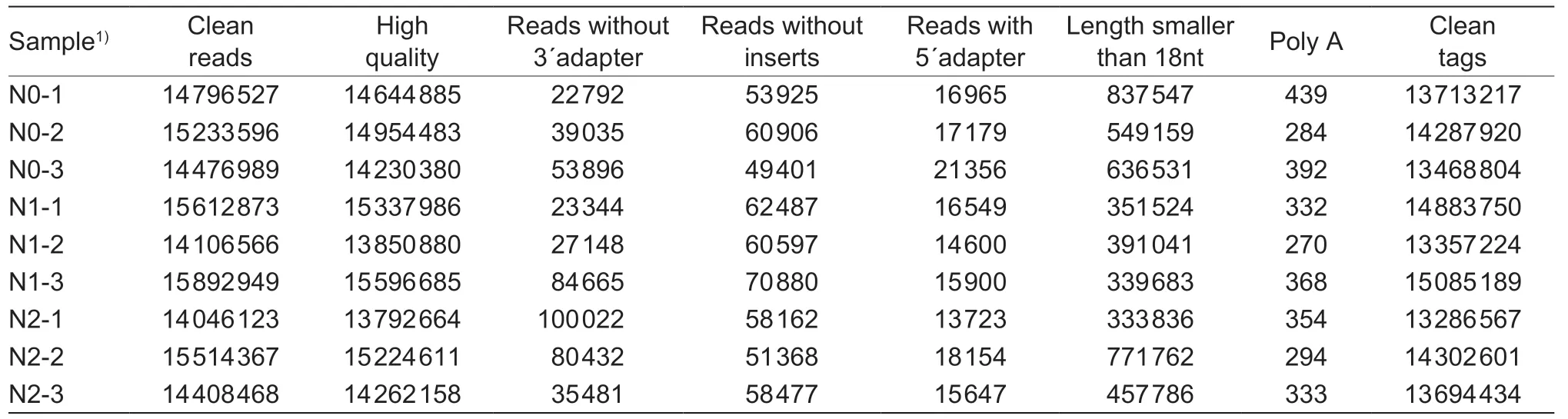
Table 1 Summary of mRNA sequencing datasets
A total of 2 997 DEGs were obtained in group N0vs.N1,where 1 123 up-regulated DEGs and 1 874 downregulated DEGs were identified; 3 330 DEGs were identified in group N0vs.N2,of which 1 389 were upregulated,and 1 941 were down-regulated (Fig.3-A).In general,the number of DEMIs increased with the N application,exactly as 225 and 297 DEMIs were upregulated,and 174 and 177 DEMIs were down-regulated under N1 and N2 compared with N0 (Fig.3-B).
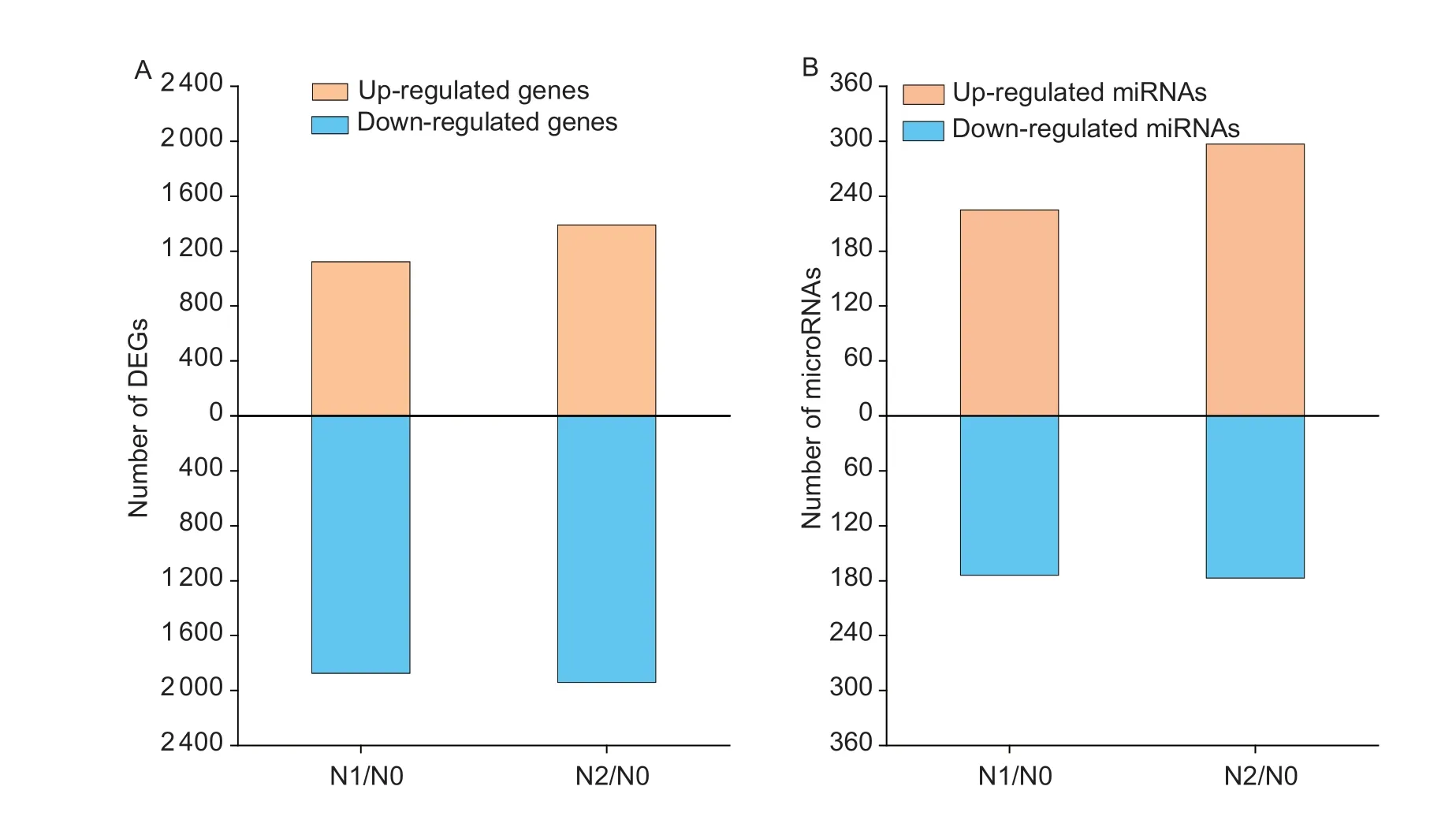
Fig.3 The expression of differentially expressed genes (DEGs,A) and differentially expressed microRNAs (B) in maize kernels under different N rates.N1/N0 and N2/N0 indicate the change of genes or microRNAs in N1 or N2 compared with that in N0.N0,no N fertilizer; N1 and N2 indicate 100 and 200 kg N ha-1,respectively.
3.3.Functional analysis of DEGs
To explore the function of DEGs under different N rates,the 4 095 DEGs were divided into eight trends.Among these trends,four were significantly shown,including 406 DEGs in profile 0,1 895 DEGs in profile 1,1 242 DEGs in profile 6,and 350 DEGs in profile 7.We then chose the DEGs in the profiles for KEGG pathway analysis (Fig.4).
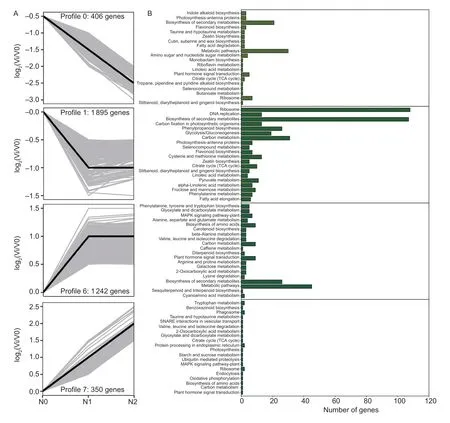
Fig.4 Differentially expressed genes and Kyoto encyclopedia of genes and genomes (KEGG) pathways under different N rates in maize kernels.A,gene expression patterns under different N rates.The gray and black lines represent each gene’s expression pattern and the expression trend of all genes,respectively.Vi,N1 or N2; V0,N0.B,KEGG enrichment analysis of four significant clusters under different N rates.N0,no N fertilizer; N1,N2,and N3 indicate 100,200,and 300 kg N ha-1,respectively.
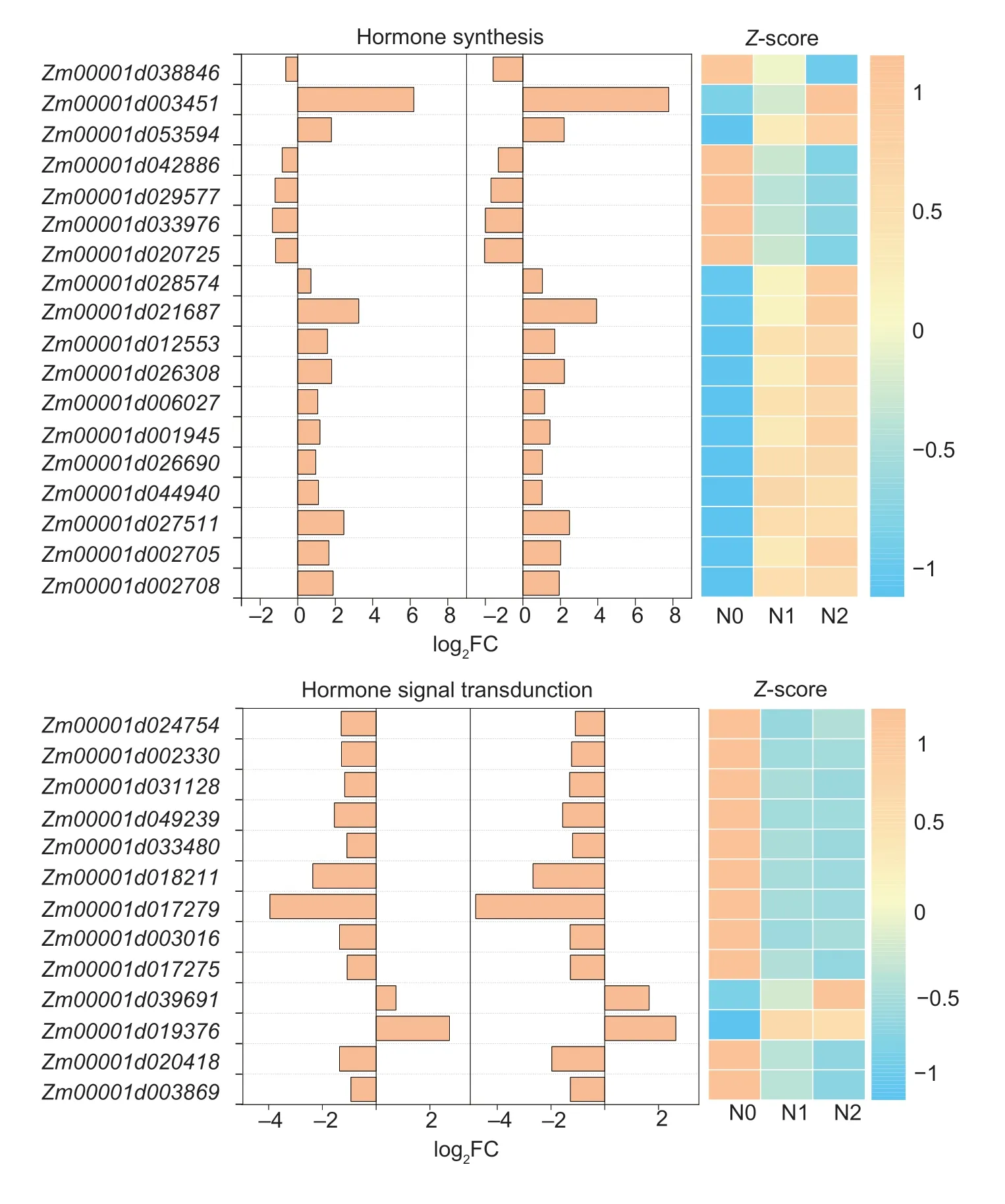
Fig.5 Kyoto encyclopedia of genes and genomes involved in hormone synthesis and signal transduction under different N rates in maize kernels.N1/N0 and N2/N0 indicate the change of genes in N1 or N2 compared with N0.log2FC,log2fold changes.N0,no N fertilizer; N1 and N2 indicate 100 and 200 kg N ha-1,respectively.
In profile 0,the DEGs were down-regulated with the increase of N application.These DEGs were mainly enriched in “biosynthesis of secondary metabolites” and “metabolic pathways”,“ribosome”,and “plant hormone signal transduction”.The DEGs in profile 1 were downregulated from N0 to N1 but were no longer downregulated when the N rate exceeded N1.In profiles 6 and 7,with the increase of N,the DEGs were upregulated,or they were first up-regulated and then remained unchanged.Interestingly,“plant hormone signal transduction” took part in profiles 0,6,and 7,which attracted attention.In addition,the “MAPK signaling pathway” plays an important role in hormone signal transduction.The DEGs of “cysteine and methionine metabolism”,“phenylpropanoid biosynthesis”,“tryptophan metabolism”,“carotenoid biosynthesis”,and “zeatin biosynthesis” involved in plant hormone signal transduction were also analyzed.Finally,we obtained 31 DEGs in maize kernels under different N rates.
3.4.Analyses of DEGs involved in plant hormone syntheses and signal transduction
In the IAA pathway (Figs.5 and 6),N1 and N2 upregulatedZm00001d039691(TAA1,tryptophan aminotransferase related),which is involved in IAA synthesis.In addition,N down-regulated two genes in IAA signal transduction,includingZm00001d033976(AUX/IAA,auxin-responsive protein) andZm00001d029577(GH3,auxin-responsive GH3 gene family).N upregulatedZm00001d001945(ARF) andZm00001d026308(SAUR,SAUR family protein) expression.
In the ABA pathway (Figs.5 and 6),the N application up-regulatedZm00001d019376expression,which encodes abscisic-aldehyde oxidase (AAO3).Three genes (Zm00001d042886,Zm00001d038846,andZm00001d026690) related to protein phosphatase 2C (PP2C) and serine/threonine-protein kinase SRK2 (SnRK2) were up-regulated in N1 and N2.However,Zm00001d028574(PP2C) was down-regulated in N1 and N2.
In the CTK pathway (Figs.5 and 6),the cytokinin trans-hydroxylase (CYP735A) gene (Zm00001d020418) and the adenylate dimethylallyltransferase (IPT) gene (Zm00001d003869) were down-regulated in N1 and N2.
Ent-copalyl diphosphate synthase (Zm00001d032961) and gibberellin 2beta-dioxygenase (Zm00001d024175) were up-regulated in N1 and N2 in the GA pathway (Figs.5 and 6).In the ETH pathway,seven genes were obtained in our study (Figs.5 and 6).Only one gene(Zm00001d003451,EIN3,ethylene-insensitive protein 3)was up-regulated.Furthermore,six genes were downregulatedin N1 and N2:Zm00001d024754(met),Zm00001d002330(DNMT1,DNA methyl transferase 5),Zm00001d031128 (met E),Zm00001d049239(ahc Y,adenosylhomocysteinehydrolase1),Zm00001d033480(met),andZm00001d018211(ACO,1-aminocyclopropane-1-carboxylate oxidase 35).
In the salicylic acid pathway,the genes (Zm00001d017279,Zm00001d003016,andZm00001d017275) in salicylic acid synthesis were down-regulated; however,the genes (Zm00001d006027andZm00001d012553) in salicylic acid signal transduction were upregulated.Only one gene (Zm00001d020725),which encodes BRI1 kinase inhibitor 1 (BKI1),was obtained in the brassinolide signal transduction pathway.
Results showed that IAA,CTK,GA,ABA,ETH,BR,JA,and SA have important roles in grain development (Fig.6).TAA was involved in the tryptophan metabolism and auxin synthesis,and the TAA gene was up-regulated in N2 and N1 relative to N0.The genes encoded AUX/IAA and GH3 expressed lower in N2 and N1 compared with N0; however,the genes encoded ARF and SAUR were up-regulated after N application.In the ABA synthesis and signal transduction pathway,the majority of these genes were expressed higher in N2 and N1 relative to N0,except for the genes related to PP2C.The N application also affected the synthesis of CTK and GA and the signal conduction of BR.Interestingly,the genes related to the synthesis of ETH and SA were downregulated,while the genes involved in the signal conduction of ETH and SA were up-regulated after N supply.
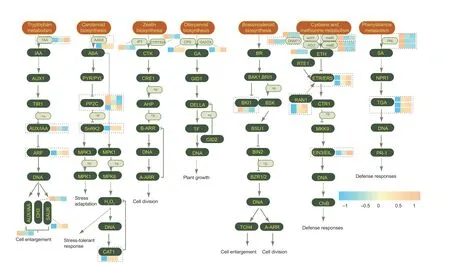
Fig.6 The synthesis and transduction of hormones in maize kernels under different N rates.The three rectangles in a group are N1,N2,and N3 from left to right,and the different color of the rectangle indicates the different Z-score value.N1,N2,and N3 indicate 100,200,and 300 kg N ha-1,respectively.IAA,indole-3-acetic acid; ABA,abscisic acid; CTK,cytokinin; GA,gibberellin; BR,brassinosteroid; ETH,ethylene; SA,salicylicacid.
3.5.Construction of the co-expression network between DEMIs and DEGs
With the help of Cytoscape,we used the DEGs screened in hormone synthesis and signal transduction pathway and DEMIs to establish a regulatory network diagram (Fig.7).In the diagram,we used the DEGs as the core and DEMIs as the target to reveal how microRNAs affect the gene expression under different N rates.In some cases,the interaction network showed that a single microRNA could be targeted by various mRNAs,whereas a single mRNA could also target different microRNAs.A total of 9 DEGs were regulated by 14 microRNAs,which formed 26 pairs.Moreover,12 microRNAs had predicted one gene,and one gene had 15 predicted microRNA-binding sites.
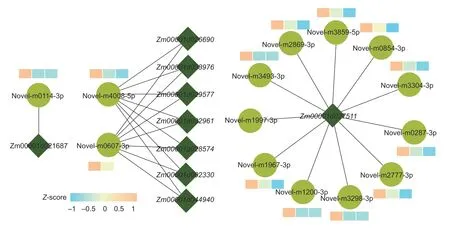
Fig.7 microRNA-mRNA correlation network.Circles represent microRNA,diamonds represent genes,and the three rectangles in a group are N1,N2,and N3 from left to right.The different colors of the rectangle indicates the different Z-score value.N1,N2,and N3 indicate 100,200,and 300 kg N ha-1,respectively.
3.6.DEGs were verified by quantitative real-time PCR
qRT-PCR was used to verify the reliability of our transcriptome data.We selected 22 genes from the DEGs,and the results showed that the expression patterns of these genes at 0,100,and 200 kg N ha–1showed similar trends to our RNA-seq results,which proved the accuracy of RNA-seq analysis (Fig.8).
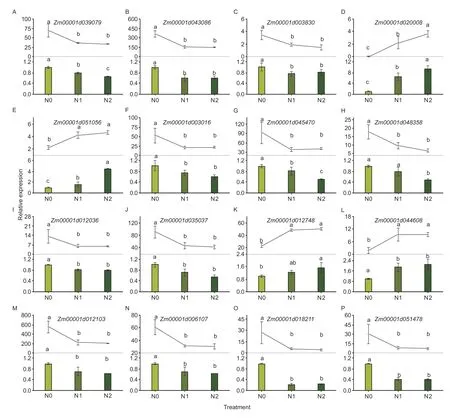
Fig.8 qRT-PCR analysis of differentially expressed genes in maize grains under different N rates.N0,no N fertilizer; N1 and N2 indicate 100 and 200 kg N ha-1,respectively.Bars mean SD (n=3).Different lowercase letters indicate the difference at P<0.05.Broken lines represent RNA-seq,and bars represent qRT-PCR.
4.Discussion
4.1.Changes in maize grain weight under different N rates
Improving the utilization efficiency of N fertilizers and increasing N in the grains cannot only increase the weight of the grains but also help to improve the grain quality.One method to improve N utilization efficiency is to reduce the input of N fertilizer without reducing grain weight (Goodetal.2004; Lietal.2021).In this experiment,N applications of 100,200,and 300 kg ha–1significantly increased the grain weight compared with no N application,but N applications higher than 200 kg ha–1did not significantly increase the grain weight,showing that increasing the input of N cannot effectively promote the development of grains.
4.2.Regulation of hormone contents in grain develop- ment
Hormones play an important regulatory role in grain development.Numerous studies have shown that N can regulate grain development by affecting hormones (Liuetal.2010,2013; Yuetal.2017; Weietal.2019; Caoetal.2020; Yueetal.2022).Our experiment found that the dry weight of 100 kernels was positively correlated with the endogenous contents of various hormones (except for ETH) under different N rates,which showed the role of hormones in the grain-filling process (Lietal.2019).The high kernel’s weight under N2 and N3 might be because high IAA,GA,and CTK contents promoted endosperm cell division,and high ABA content facilitated the accumulation of photosynthetic products (Wangetal.2006; Zhangetal.2017).We observed high kernel weight and reduced EHT content under N2 and N3,which showed that high ETH content under the N deficiency condition decreased kernel weight by inhibiting grain filling (Yinetal.2017).Liuetal.(2008) found that maize grain filling is regulated at the molecular level,and hormone signaling plays an important role in this process.Jinetal.(2015) found that small RNAs are involved in the regulation of hormonal signaling in maize grain filling.In the present study,we comprehensively analyzed the expression of microRNAs and mRNAs in maize grains.We identified 1 123 and 1 389 up-regulated DEGs and 1 874 and 1 941 down-regulated DEGs in N0vs.N1 and N0vs.N2 groups.In addition,33 DEGs were obtained in hormone synthesis and signal transduction through KEGG pathways.This determined that N regulates grain filling at the molecular level,while hormones take part in maize grain filling under different N applications.Studies have shown that small RNAs are involved in the development of crop grains (Li et al.2016; Zeng et al.2019; Zhang et al.2019; Hou et al.2020; Yang et al.2021).Therefore,it is necessary to analyze the regulation of small RNAs on maize grain development under different N application conditions.Presently,we obtained 399 and 474 DEMIs in groups N1 vs.N0 and N2 vs.N1,respectively.Remarkably,14 novel DEMIs regulated the genes involved in hormone synthesis and transmission,and it would be interesting to study their roles and functions in hormones to regulate grain filling.
4.3.Integration analysis of microRNAs and mRNAs in IAA pathway
Zm00001d039691encoding TAA (tryptophan aminotransferase) was up-regulated by N application and participated in auxin biosynthesis through tryptophan metabolism (Yueetal.2014).Ultimately,the gene overexpression led to IAA accumulation at the physiological level,which is similar to the physiological indicators we observed.The IAA accumulation promoted the degradation of auxin/indoleacetic acid proteins (AUX/IAA) and then released ARF,activating downstream gene expression to cope with environmental stress and regulate plant growth (Grayetal.2011).The genesZm00001d033976(Aux/IAA) andZm00001d029577(GH3) were down-regulated,andZm00001d001945(ARF) andZm00001d026308(SAUR) were up-regulated by N application.This indicated that N application was more favorable to activate downstream genes in response to environmental stress.Gretchen Hagen 3 (GH3) gene family plays an important role in coping with environmental stress and responding to various biotic stress (Kumaretal.2012).In this experiment,N application down-regulated genes related to GH3,and the result was similar to Effahetal.(2022).It has to be considered that GH3 is also regulated by fruit senescence (Kumaretal.2012).High expression of GH3 under no N application may lead to premature grain maturity,which is not conducive to grain filling.In addition,our results also further support the assertion that N application regulated maize growth through affectedSAURexpression (Wangetal.2019).Interestingly,the genes encoding Aux/IAA,GH3,and ARF were regulated by novel-m4008-5p and novel-m0607-3p,which are the new DEMIs.Novelm4008-5p and novel-m0607-3p may take part in maize grain development under different N rates by regulating hormones.
4.4.The microRNAs and mRNAs involved in ABA pathway
Under N application,an ABA synthesis-related gene (AAO3,Zm00001d018211) was up-regulated,which might have led to an increase in ABA content.The ABA signaling network operates in a manner where the recipient of ABA by PYR/PYL can inhibit the activity of type 2C protein phosphatases (PP2Cs) and release SnRK2,thereby activating downstream factors to regulate crop growth (Umezawaetal.2009).In this experiment,four genes were involved in the ABA signaling core pathway PYR/PYL-PP2C-SnRK.Among these genes related to PP2Cs,two genes (Zm00001d042886andZm00001d038846) were down-regulated,while one gene (Zm00001d028574) was up-regulated.Zm00001d028574was negatively regulated by novel-m4008-5p and novel-m0607-3p.However,Zm00001d042886andZm00001d038846were not regulated by microRNA in our study.In addition,the genes encoding SnRK2 were up-regulated,and the gene increased with the increase in N application.Finally,ABA signal transduction was enhanced by increasing catalase (CAT) activity and eliminating hydrogen peroxide in maize.Interestingly,we found 11 novel microRNAs with a negative regulatory effect on CAT.The result indicates that the 11 novel microRNAs may participate in maize’s response to abiotic stress caused by N stress.
4.5.The mRNAs in other hormonal pathways
N application increased the expression of genes related to GA synthesis,a multifunctional plant signal that plays an important role in grain development (Bari and Jones 2009).In addition,N has little effect on BR.ETH has a great influence on the late growth stage of crops,which is related to the regulation of crop senescence (Burg and Burg 1965).In our study,six genes (Zm00001d024754,Zm00001d002330,Zm00001d031128,Zm00001d049239,Zm00001d033480,andZm00001d018211) related to ETH synthesis were down-regulated by N application,and these genes were closely related to ETH synthesis.ACOexpression affects 1-aminocyclopropane-1-carboxylate (ACC) content,which is the precursor of ETH (Mouetal.2020).Recently,Ningetal.(2021) found that the lower expression of theACO2gene can lead to an increase in maize grain weight and yield.In other words,theACO2gene has a negative correlation with corn grain weight.This result is similar to ours: N application decreasedACOgene expression and increased grain weight.When the external environment induces ETH production,ETH combines with ethylene receptors (ETR/ERS) to inhibit the CTR1,promoting EIN3/EIL and thus leading to an ETH response (Taoetal.2015).In this pathway,the N application up-regulated the genes (Zm00001d021687andZm00001d053594) related to ETR/ERS.Simultaneously,the ETH synthesis-related genes were down-regulated,activating CTR1.In addition,ETR/ERS is a negative regulator of fruit senescence (Mataetal.2015),so N application may delay grain maturity and is conducive to grain filling.Interestingly,EIN3 is not only the main network hub of ETH signal transduction but also integrates the signal transduction between various hormones and between hormones and stress,which forms a complex transcriptional regulation network ensuring the growth and development of plants in various complex environments (Anetal.2012; Lingametal.2011; Zhuetal.2021).More importantly,EIN3 regulates more than 1 000 genes involved in plant growth and development (Changetal.2013).In our study,the gene (Zm00001d003451) involved in EIN3/EIL was upregulated by N application.However,the regulation of grain development by EIN3 needs to be further explored because the functions of EIN3 are diverse.Our study showed that the gene expression related to SA synthesis decreased with increasing N application rate,which was similar to the findings of Effahetal.(2022).
5.Conclusion
This study proved that N regulates hormone synthesis and signal transduction at the transcriptome level,affecting grain development.At the same time,we identified the novel microRNAs that mediate hormonal responses to nitrogen in maize kernels.We gain new insights into hormone regulation of grain development.This knowledge broadens the understanding of signaling pathways and may reveal regulators in maize kernels that respond to nitrogen stress.Further elucidation of the hormonal pathway may contribute to novel pathways for improving maize kernel weight under low nitrogen conditions.
Acknowledgements
This research was supported by the Major Special Research Projects in Gansu Province,China (22ZD6NA009),the State Key Laboratory of Aridland Crop Science,Gansu Agricultural University,China (GSCS-2022-Z02),the National Natural Science Foundation of China (32260549),and the National Key R&D Program of China (2022YFD1900300).We appreciate assistance in the field and laboratory by students of the Rainfed Agricultural Experimental Station of Gansu Agricultural University.
Declaration of competing interest
The authors declare that they have no conflict of interest.
杂志排行

Journal of Integrative Agriculture的其它文章
- Comparison of cell wall changes of two different types of apple cultivars during fruit development and ripening
- Colonization by Klebsiella variicola FH-1 stimulates soybean growth and alleviates the stress of Sclerotinia sclerotiorum
- Degradation effects on dichlorvos by a biocontrol strain,Trichoderma atroviride T23
- Novel 18β-glycyrrhetinic acid amide derivatives show dual-acting capabilities for controlling plant bacterial diseases through ROSmediated antibacterial efficiency and activating plant defense responses
- Effects of methionine treatment on storage quality and antioxidant activity of postharvest jujube fruit
- Physicochemical properties and antibacterial mechanism of theabrownins prepared from different catechins catalyzed by polyphenol oxidase and peroxidase