UHPLC-MS/MS法测定依那普利氢氯噻嗪复方制剂中潜在基因毒性杂质
2023-09-10袁松张才煜庾莉菊陈华张庆生
袁松,张才煜,庾莉菊,陈华,张庆生
(中国食品药品检定研究院,国家药品监督管理局化学药品质量研究与评价重点实验室,北京 102629)
基因毒性杂质是指在很低的浓度下即可诱导基因突变并导致染色体的断裂和重排的杂质,具有潜在的致癌性[1]。N-亚硝胺类化合物由于在细胞色素P450酶介导代谢活化后,生成不稳定的α-羟甲基-N-亚硝胺,最终形成的高度亲电物质与DNA反应形成烷基化的DNA碱基对,从而具有很强的致癌性[2-3],ICH M7(R1)指南[4]已将N-亚硝胺类化合物列入“关注队列”。近年来多个品种的药品中检测出了N-亚硝胺类化合物,引起了各国药品监管机构的广泛关注,出台了一系列N-亚硝胺杂质的控制方案,并要求申请人评估存在N-亚硝胺杂质的风险[5-7]。
氢氯噻嗪(hydrochlorothiazide,HZCT)作为一种噻嗪类利尿药,在临床上广泛应用于治疗水肿和高血压,不论是单用还是与其他降压药联用都疗效明确,尤其适用于老年高血压、单纯收缩期高血压或伴心力衰竭患者,也是难治性高血压的基础用药之一[8]。氢氯噻嗪结构(见图1)中存在2个环状仲胺结构,由于其存在弱碱性的芳香环-烷基氨基,在强酸性条件下,氢氯噻嗪极易亚硝基化生成N-亚硝基氢氯噻嗪(4-nitrosohydrochlorothiazide,NO-HZCT),其结构见图1,Gold等[9]的研究显示HZCT的亚硝化只发生在芳香环-烷基氨基位点,并且与亚硝酸具有一级反应动力学,在极其微量亚硝酸盐的存在下,氢氯噻嗪有生成基因毒性杂质NO-HZCT可能,因此急需开发一种灵敏度高且专属性强的定量方法对相关制剂中可能存在的NO-HZCT进行检测,以保证药品安全。
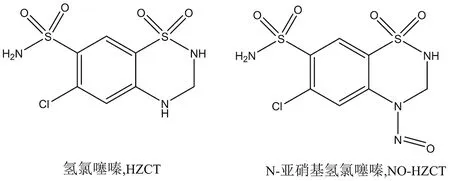
图1 氢氯噻嗪与N-亚硝基氢氯噻嗪化学结构
由于基因毒性杂质对检测灵敏度有着很高的要求,文献中N-亚硝基杂质的检测方法以超高效液相色谱-串联质谱(UHPLC-MS/MS)[10-13]或气相色谱-串联质谱(GC-MS/MS)[14-16]为主。目前,尚无NO-HZCT杂质检测方法的报道。本研究拟参考文献方法合成NO-HZCT[9],并建立UHPLC-MS/MS方法以检测依那普利氢氯噻嗪片和依那普利氢氯噻嗪分散片中NO-HZCT含量,为质量控制提供参考。
1 材料
1.1 仪器Agilent 1290 Infinity II-6470QQQ液质联用仪(美国Agilent公司),配有电喷雾离子源(ESI)以及MassHunter数据处理系统;Thermo Ultimate3000-Orbitrap120液相色谱-静电场轨道肼质谱联用仪(美国Thermo公司),配有电喷雾离子源(H-ESI);XP205DR电子天平(0.01 mg,瑞士Mettler Toledo公司);XPE26电子天平(0.001 mg,瑞士Mettler Toledo公司); VD53真空干燥箱(德国Binder公司)。
1.2 试药与试剂乙腈(质谱级、中国迪科马科技有限公司);甲酸(质谱级、德国Merck公司);甲酸铵(质谱级,德国Merck公司);水为超纯水;乙酸、高氯酸、亚硝酸钠均为分析纯,购于国药集团有限公司;氢氯噻嗪原料药(批号:H181007-1,常州制药厂有限公司);依那普利氢氯噻嗪分散片、依那普利氢氯噻嗪片(Ⅰ)、依那普利氢氯噻嗪片(Ⅱ)均为市场采购。
2 方法与结果
2.1 NO-HZCT的合成称取氢氯噻嗪原料药1.183 g,加入25%乙酸溶液80 mL,振摇使混匀,用高氯酸调节pH值为1.022,缓慢加入亚硝酸钠0.654 g,边加边振摇,以800 r·min-1搅拌90 min,过滤,取沉淀60 ℃减压干燥4 h,即得淡黄色粉末1.132 4 g,得率为95.7%。采用高分辨质谱(见图2A)和核磁(见图2B、2C)对其结构进行了确证,并采用核磁定量测得含量为94.7%。

图2 结构确证-高分辨质谱图(A,H-ESI-)、核磁共振氢谱(B)和碳谱(C)
2.2 溶液制备
2.2.1 对照品溶液配制精密称取NO-HZCT对照品10.70 mg,置100 mL量瓶中,加乙腈 溶解并稀释至刻度,摇匀,作为对照品储备液;精密量取对照品储备液0.1 mL,置100 mL量瓶中,用乙腈稀释至刻度,摇匀,作为100 ng·mL-1的溶液,精密量取1.0、1.0、2.0、5.0、10.0、10.0、10.0 mL,分别置200、100、100、100、100、50、20 mL量瓶中,用乙腈稀释至刻度,摇匀,制成每1 mL中分别约含NO-HZCT 0.5、1、2、5、10、20、50 ng的对照品溶液,作为系列线性溶液。
2.2.2 供试品溶液制备取依那普利氢氯噻嗪片(Ⅰ)、依那普利氢氯噻嗪片(Ⅱ)或依那普利氢氯噻嗪分散片20片,精密称定,研细,精密称取约相当于氢氯噻嗪25 mg的细粉,置15 mL离心管中,精密加入乙腈5 mL,涡旋5 min,滤过,取续滤液作为供试品溶液。
2.3 色谱及质谱条件
2.3.1 色谱条件采用Agilent Eclipse Plus C18RRHD(3.0 mm×150 mm,1.8 μm)色谱柱,以10 mmol·L-1甲酸铵-0.1%甲酸的水溶液作为流动相A,以0.1%甲酸的乙腈溶液作为流动相B,梯度洗脱:0.0→7.0 min,20% B~30% B;7.0→7.1 min,30% B~20% B;7.1→10.0 min,20% B。体积流量0.6 mL·min-1,柱温40 ℃,进样盘温度为5 ℃,进样体积5 μL。
2.3.2 质谱条件采用电喷雾离子源(Electrospray Ionization Source,ESI),负离子扫描模式,干燥气温度300 ℃,干燥气体积流量6 L·min-1,雾化气压力310.275 kPa,鞘气温度325 ℃,电离电压500 V。采用多反应监测(multiple reaction monitor,MRM)模式,以m/z324.9→280.9作为定量离子对,碎裂电压为70 V,碰撞能量为8 V,m/z324.9→293.9作为定性离子对,碎裂电压为70 V,碰撞能量为28 V。
2.4 方法学考察
2.4.1 专属性试验取乙腈、“2.2.1”项下5 ng·mL-1的对照品溶液和供试品溶液按“2.3”项下条件分别进样检测,记录色谱图(见图3),结果显示,在所建立的色谱-质谱条件下NO-HZCT的保留时间约为4.6 min,主成分依那普利氢氯噻嗪和乙腈对其检测均没有干扰。
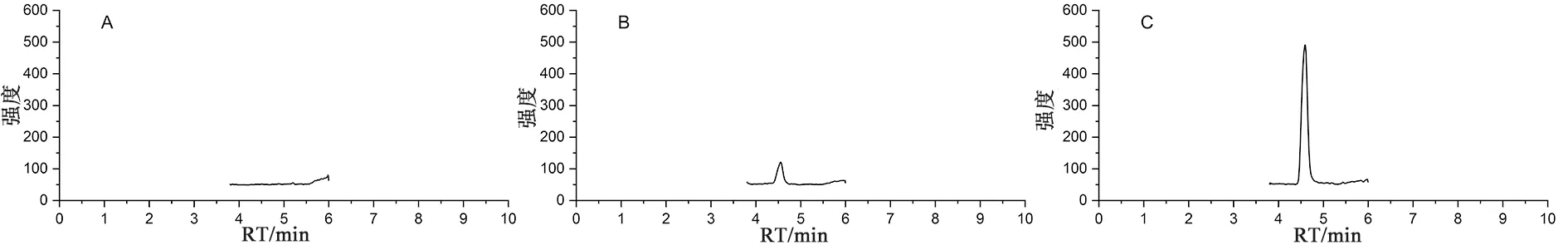
图3 乙腈(A)、对照品溶液(B)和供试品溶液(C)提取离子流色谱图
2.4.2 检测限与定量限取“2.2.1”项下0.5 ng·mL-1的对照品溶液,用乙腈逐级稀释,直至NO-HZCT峰的信噪比(S/N)约为3时的质量浓度作为检测限(LOD),S/N约为10时作为定量限(LOQ),测得NO-HZCT的检测限与定量限分别为0.08 ng·mL-1和0.27 ng·mL-1。
2.4.3 线性关系考察分别取“2.2.1”项下各质量浓度的系列线性溶液,按 “2.3”项条件进样检测,并记录峰面积,以NO-HZCT质量浓度为横坐标(X),峰面积为纵坐标(Y)进行线性回归,得线性方程为Y=124.07X-1.16,相关系数(r)为0.999 8,结果表明NO-HZCT杂质质量浓度0.51~50.67 ng·mL-1范围内与峰面积呈良好的线性关系。
2.4.4 系统精密度取“2.2.1”项下1 ng·mL-1的对照品溶液,按 “2.3”项条件连续进样6次,记录峰面积,计算得NO-HZCT峰面积的相对标准偏差(RSD)为4.43%,结果表明系统精密度良好。
2.4.5 重复性取依那普利氢氯噻嗪片(Ⅰ),按“2.2.2”项下方法平行制备6份供试品溶液,照“2.3”项下色谱条件进样检测,记录峰面积,按外标法计算6份供试品溶液中NO-HZCT含量的RSD为3.31%,表明方法具有良好的重复性。
2.4.6 加样回收率试验精密称取依那普利氢氯噻嗪片(Ⅰ)细粉9份(约相当于氢氯噻嗪 25 mg),置15 mL量瓶中,精密加入“2.2.1”项下制备的5、10、20 ng·mL-1的对照品溶液5 mL,照“2.2.2”项下方法制备溶液,低、中、高3个质量浓度点的供试品各3份,按照“2.3”项下色谱条件进样检测,结果见表1,低、中、高3个浓度点的回收率分别为 93.10%(RSD3.7%,n=3)、104.30%(RSD1.0%,n=3)和106.48%(RSD1.8%,n=3 ),结果表明方法回收率良好。
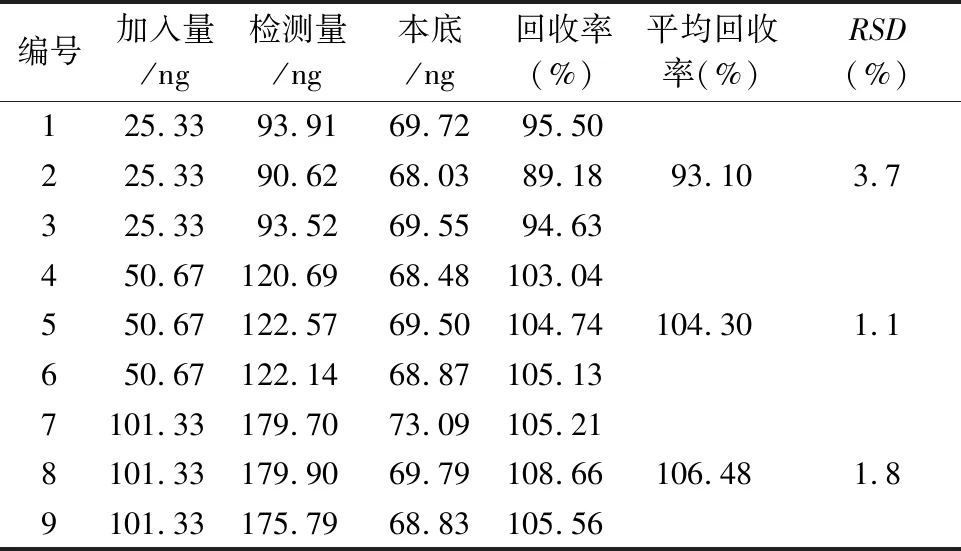
表1 加样回收率结果
2.4.7 溶液稳定性取“2.2.1”项下5 ng·mL-1的对照品溶液,放置于进样盘(5 ℃),样品溶液检测完毕后再次进样检测,0 h和5 h时NO-HZCT峰面积的相对偏差为3.75%,结果表明在5 ℃条件下,对照品溶液5 h内稳定。
2.4.8 提取效率考察取依那普利氢氯噻嗪片(I),按“2.2.2”项下方法平行制备2份供试品溶液,分别涡旋5 min和10 min,照“2.3”项下色谱条件进样检测,计算得两份供试品溶液中NO-HZCT含量的相对偏差为2.07%,结果表明涡旋5 min即可提取完全,为提高实验效率,将涡旋时间设定为5 min。
2.5 样品测定取依那普利氢氯噻嗪片(Ⅰ)、依那普利氢氯噻嗪片(Ⅱ)或依那普利氢氯噻嗪分散片20片,精密称定,研细,精密称取约相当于氢氯噻嗪25 mg的细粉,置15 mL离心管中,精密加入乙腈5 mL,涡旋5 min,滤过,取续滤液作为供试品溶液。测得依那普利氢氯噻嗪分散片,依那普利氢氯噻嗪片(Ⅰ),依那普利氢氯噻嗪片(Ⅱ)中NO-HZCT均有检出。
3 讨论
本研究初期首先对样品溶解/提取的溶剂进行了考察,由于流动相为水-乙腈体系,且NO-HZCT在水中溶解性较差,为避免溶剂效应,选择以乙腈溶解后用水稀释至刻度,但在考察中发现,稀释剂中加入水-乙腈溶剂后(水-乙腈=80∶20),NO-HZCT极其不稳定,并且其降解速率与水的pH值有关。以pH 7.0的磷酸盐缓冲液-乙腈(80∶20)为稀释剂时,在25 ℃条件下,NO-HZCT经过30 min后降解约60%(按峰面积计),180 min时几乎降解完全;以pH 2.0的磷酸盐缓冲液-乙腈(80∶20)为稀释剂时,在25 ℃条件下,NO-HZCT经过30 min后降解约47%(按峰面积计),180 min时降解约80%,且其主要降解杂质为氢氯噻嗪;以纯乙腈作为稀释剂时NO-HCZT在5 h内稳定,故本次试验以乙腈作为稀释剂对色谱-质谱条件进行优化。后续将对NO-HZCT在水中的降解情况进行进一步的深入研究。
NO-HZCT为N-亚硝基类化合物,属于ICH M7(R1)指南中“关注队列”的遗传毒性杂质,需对其限度进行严格控制。目前尚没有N-亚硝基氢氯噻嗪的毒理学数据,暂无明确数据,还需对其生理毒性进行进一步研究。检测结果提示应在生产和存储过程中加强监控,并进行安全风险评估,建立适当的控制策略,保证药品安全。
4 结论
本研究通过合成氢氯噻嗪中潜在基因毒性杂质N-亚硝基氢氯噻嗪,首次建立了依那普利氢氯噻嗪复方制剂中N-亚硝基氢氯噻嗪的超高效液相-串联质谱的检测方法,该方法灵敏度高、专属性强,可准确地对依那普利氢氯噻嗪复方制剂中基因毒性杂质N-亚硝基氢氯噻嗪进行定量检测,可为依那普利氢氯噻嗪制剂的质量控制提供参考,保障药品质量安全。
