欧盟与我国GMP无菌药品附录差异分析与探讨
2023-09-10胡敬峰明奕王金子宋凯樊红延冯巧巧
胡敬峰,明奕,王金子,宋凯,樊红延,冯巧巧
(1.山东省食品药品审评查验中心,山东 济南 250014;2.国家药品监督管理局高级研修学院,北京 100073)
欧盟《人用和兽用药品生产质量管理规范指南》附录1“无菌药品生产”(以下简称“欧盟GMP附录1”)于2022年8月25日正式发布,这是欧盟2015年发布对无菌药品附录修订的概念文件草案后历经7年最终修订完成的[1]。该附录要求的最后实施期限是 2023年8月25日,需要指出的是,附录中的第8.123条“冻干机和相关产品转移和装载/卸载区域应经过设计,尽可能减少操作人员的干预”的最后实施期限为 2024年8月25日。欧盟GMP附录1首次颁布是在1971年,之后经过多次修订,上一次修订是2008年,该版本是与我国现行《药品生产质量管理规范(2010年修订)》的附录1“无菌药品”(以下简称“中国GMP附录1”)内容较为接近的版本[2-3]。
本次欧盟GMP附录1修订版从覆盖范围、章节以及内容都有非常大的调整,为首次全面修订。该附录规定了无菌药品生产质量控制的基本原则,详细说明了对无菌药品生产企业质量体系的要求,应“确保所有活动得到有效控制,以尽可能减少无菌产品中的微生物、微粒和内毒素/热原污染风险”,明确了洁净厂房、设备、人员、生产等方面的具体要求[4]。我国GMP附录1自2010年至今已有13年未修订,难以满足当今无菌药品GMP管理的要求。
2022年版欧盟GMP附录1是由世界卫生组织(WHO)、欧洲药品管理(EMA)和国际药品检查合作组织(PIC/S)联合起草,有利于全球标准的协调一致,也将对我国制药工业的发展产生积极而又深远的影响。学习、研究和有效转化应用这一附录,将对我国制药工业创新发展和建立国际化竞争优势发挥重要促进作用。
为了解国际无菌药品生产质量管理的最新发展趋势与要求,把握我国无菌药品质量管理的现状,促进我国无菌药品质量提升,本文对比分析了欧盟与我国GMP附录1的主要差异点,以期为我国GMP附录1的修订和我国无菌药品生产企业质量提升提供参考。
1 整体性差异
2022年版欧盟GMP附录1共11章,295条,包括范围、原则、药品质量体系、厂房、设备、公用设施、人员、生产和具体技术、环境监测和工艺监测、质量控制、术语等[5-6]。中国GMP附录1共15章,81条,包括范围、原则、洁净度级别及监测、隔离操作技术、吹灌封技术、人员、厂房、设备、消毒、生产管理、灭菌工艺、灭菌方法、无菌药品的最终处理、质量控制、术语等。
2008年版欧盟GMP附录1仅有17页126条,而2022年版则长达59页。2022年版本引入了范围、药品质量体系(pharmaceutical quality system,PQS)、公用设施、环境和过程监测、术语表等新的章节,同时加强了质量风险管理、污染控制策略、新技术和新工艺等结构化内容的指导,在内容上有了大幅增加,各项规定更加翔实和具体。2022年版文件还包含一些中国GMP附录1没有规定的内容,如:污染控制策略、药品质量体系、公用设施、生产和具体技术(成型-灌装-密封技术、冻干、密闭系统、一次性系统)等。各版本无菌药品附录整体性差异对比详见表1。

表1 欧盟与我国GMP无菌药品附录整体性差异对比
2 差异分析
欧盟与中国GMP附录1最主要的差异集中在欧盟附录1的拓展了适用范围,明确了新的概念(如质量风险管理、污染控制策略、药品质量体系等),强调了新的技术(如屏障技术),提高了部分技术标准(如气流流型测试、过滤器完整性测试等),本部分对两者的主要差异点进行分析。
2.1 无菌药品与无菌产品2022年版欧盟GMP附录1在范围章节中规定“无菌产品的生产涵盖多种无菌产品类型(原料药,辅料,内包装材料和成品制剂),包装规格(从单剂量到多剂量〉,工艺(从高度自动化系统到手动工艺)和技术(如生物技术,传统小分子生产系统和密闭系统)”。中国GMP附录1中规定“无菌药品是指法定药品标准中列有无菌检查项目的制剂和原料药,包括无菌制剂和无菌原料药”。欧盟GMP附录1在适用范围方面从无菌药品拓展到无菌产品,除适用于药品外还适用于辅料、内包装材料等产品。
2.2 质量风险管理(QRM)和污染控制策略(CCS)2022年版欧盟GMP附录1范围和通则章节中提出了无菌产品生产中的质量风险管理(quality risk management,QRM)和污染控制策略(contamination control strategy,CCS)这两个在中国GMP附录1中没有涉及的概念。QRM的原则在第一章“范围”和第二章“原则”中均有描述。QRM作为一种识别、科学评估及控制质量潜在风险的前瞻性方法,应贯穿无菌药品生产全过程,包括设施、设备、系统和规程的设计和控制的过程。CCS是指针对微生物、内毒素/热原和微粒的一系列有计划的控制措施,源于现有产品和工艺的理解并确保工艺性能和产品质量。CCS的控制措施可包括:原料药和制剂的物料和组分相关的参数和属性、厂房设施设备的操作条件、中间过程控制、成品质量标准以及相关方法和监控频次等。CCS是本次附录修订新提出的概念,在第二章“原则”中进行了重点阐述,全文出现多次,明确需要在整个生产过程和所有关键控制点建立这一概念的原则。从现状来看,药品生产企业制订的文件中已经包含了许多CCS要素,但没有形成系统化的文件,欧盟GMP附录1要求CCS应为一个多元素、正式记录的文件。CCS概念的提出说明对无菌产品生产污染的控制需要全生命周期的管理:从项目的需求分析入手识别各维度污染风险;设计污染控制策略、配套控制措施来控制风险;在生产过程中进行污染的控制应对及做好日常监测;定期回顾评价污染控制策略的准确性和适应性,及时进行动态调整修正。
2.3 药品质量体系(PQS)2022年版欧盟GMP附录1使用一个章节的篇幅(第3章 药品质量体系)强调适用于无菌产品的PQS的具体要求。中国GMP在总则中规定企业应当建立药品质量管理体系,在附录1中未针对无菌药品提出药品质量体系的具体要求。欧盟GMP附录1强调了无菌药品生产的PQS应确保产品全生命周期的风险管理,对生产商、高层管理人员、负责无菌产品认证/放行的人员提出了具体要求,要对失败进行根本原因分析、CCS的开发和维护中施用风险管理,涉及的最终处理、贮存和运输相关过程不应影响产品无菌、应在批次放行之前充分调查所有不合格等。
2.4 屏障技术2022年版欧盟GMP附录1第四章“厂房”的“屏障技术”介绍了隔离器(isolator)和限制进出隔离系统(restricted access barrier system,RABS)两种技术,并明确两者是不同的技术。欧盟GMP规定隔离器“内部工作区符合A级条件”,中国GMP规定隔离操作器内部环境为“B级(ISO 5级)或更高洁净度级别”,两者对内部环境级别要求存在差异。欧盟GMP附录1明确RABS技术为“提供一个封闭但未完全密封、符合规定的空气质量条件的环境(对于无菌工艺为A级)并使用刚性外壳和一体式手套使内部与周围洁净室环境隔开的系统”,中国GMP附录1并未涉及RABS的相关规定。此外欧盟GMP附录1对隔离器和RABS手套的灭菌、完整性测试频次进行了明确的规定,要求更为严格。隔离器和RABS技术在无菌药品的生产中应用广泛,欧盟GMP对屏障技术的规定有利于促进屏障技术的规范应用,降低技术应用中的无菌风险。
2.5 洁净室环境确认与监测2022年版欧盟GMP附录1将洁净室确认(包括分级)与动态环境监测明确区别开来,洁净室确认标准在第四章“厂房”中进行了规定,环境监测标准在第九章“环境监测和工艺监测”中进行了规定。洁净室确认包含洁净室分级、微生物污染水平确认等,洁净室分级的主要依据是空气悬浮粒子标准。中国GMP附录1未区分洁净室确认(分级)标准和环境监测标准[7]。
在空气悬浮粒子方面,欧盟GMP分级标准未规定≥5 μm悬浮粒子的A静态和动态标准以及B级静态标准,欧盟GMP环境监测限值规定≥5 μm悬浮粒子的A静态和动态现值为29个/立方米,中国GMP规定≥5 μm悬浮粒子的A静态和动态标准为20个/立方米,这一点比欧盟GMP更为严格。D级环境的动态悬浮粒子标准中国GMP为“不作规定”,欧盟GMP为“未预先确定”,要求生产商根据风险评估和适用的常规数据建立动态限度,标准制定更加科学。欧盟与中国GMP各级别空气悬浮粒子标准对比见表2。
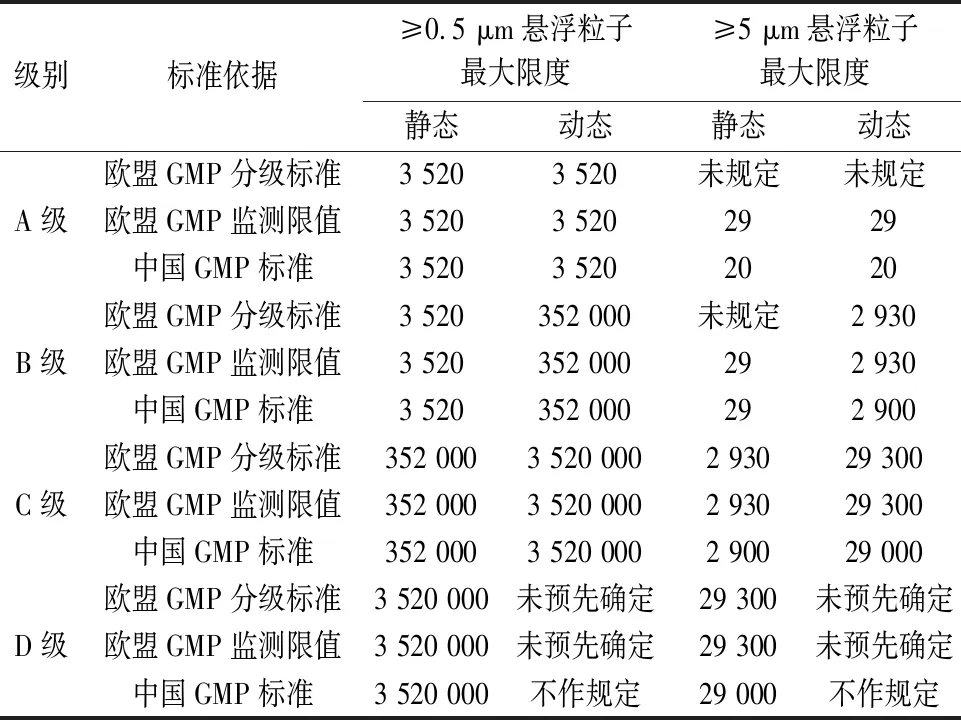
表2 欧盟与中国GMP各级别空气悬浮粒子标准对比(单位:每立方米)
在微生物限度方面,在中国GMP附录1仅规定在动态环境监测中进行微生物监测,欧盟GMP附录1要求在洁净室确认以及环境监测中均需要进行微生物监测。中国GMP规定A级的微生物监测的标准为<1 cfu,此标准在执行过程中存在如果环境监测有微生物生产,但监测结果的平均值<1 cfu,检测结果是否符合要求的争议;欧盟GMP将A级微生物标准修订为“无生产”,消除了标准的异议。欧盟与中国GMP各级别环境微生物限度标准对比见表3。

表3 欧盟与中国GMP各级别环境微生物标准对比
2.6 气流流型测试欧盟GMP附录1第四章对气流流型提出了较高的要求,条款“4.4”规定“应证明并确认整个A级区的单向流维护状态”,这就要求A级区域单向流的测试不能只测试某个位置,而应该证明整个A级区域内部的单向流是符合要求的。条款“4.15”规定“应该对洁净区和洁净室的气流流型进行可视化研究,证明空气没有从较低级别区域进入到较高级别区域,并且空气不会从不太洁净的区域(例如地板)或经过可能转移污染的操作员或设备跨越到较高级别的区域”,这就要求气流流型测试不限于A级区域,不同级别洁净区域间均需要测试,而且测试应包含静态和动态条件。中国GMP附录1仅规定“应当能够证明所用气流方式不会导致污染风险并有记录(如烟雾试验的录像)”,对具体测试要求未进行详细规定。
2.7 液体过滤器的完整性测试欧盟GMP附录1条款8.87规定“应在使用前通过完整性测试(pre-use post sterilization integrity test,PUPSIT)确认无菌过滤器组件的完整性,以检查由于使用前过滤器准备造成的损坏和完整性损失”“对液体进行除菌的除菌级过滤器,在使用后将滤器从壳体中取出之前应进行非破坏性的完整性测试”。使用前灭菌后的完整性测试(PUPSIT)要求非常高,需要避免检测时对已灭菌的过滤器系统造成二次污染。现行中国GMP附录1对于液体过滤器的完整性检测要求为“除菌过滤器使用后,必须采用适当的方法立即对其完整性进行检查并记录”,未对 PUPSIT提出要求[8-9]。
3 讨论
修订后的2022年版欧盟GMP附录1要求更加严格、内容更加全面、条款描述更加细致。①适用范围从“无菌药品”放大到“无菌产品”,涵盖了无菌原料、辅料、内包装材料以及最终成品制剂,强调无菌产品管理的目标不仅仅是微生物,还包括微粒和内毒素/热原污染;②引入了CCS的新概念,强调了QMS、PQS等理念;③对无菌生产的具体技术,如屏障技术(包括隔离器和RABS)、FFS、密闭系统、SUS等,进行了详细规定,同时引入了快速转移系统、机器人、快速微生物测试等新技术;④将洁净室确认分级与动态环境监测区别开来,各级别悬浮粒子和微生物标准设定更加科学;⑤部分内容要求比我国GMP更加严格,如气流流型测试范围、液体除菌过滤器强调使用前灭菌后完整性测试(PUPSIT)等。我国GMP附录1的基本涵盖了无菌药品生产的关键要素,但尚未明确PQS、CCS等理念,缺乏对RABS、FFS、SUS等无菌生产技术的规定。从无菌药品生产理念和无菌药品生产技术进步的角度来看,我国GMP附录1的修订有其必要性。2022年9月,PIC/S根据已经发布的2022年版欧盟GMP附录1发布了自己的GMP指南附录1,而中国已经在2021年提出加入PIC/S的申请,可以预见欧盟GMP附录1对中国制药行业的影响也会不断增强[10-11]。在国际无菌药品法规相互协同一致的背景下,我国监管部门和无菌药品生产企业要加快对欧盟GMP附录1的研究和转化,及时修订我国GMP附录1,促进我国无菌药品质量提升,建立国际化竞争优势。
