三个稀土配合物的制备、晶体结构及热分解性质
2023-09-01宗智慧周飞亚沈婧祎梁丽丽
宗智慧 周飞亚 沈婧祎 王 珊 梁丽丽
(蚌埠医学院公共基础学院 安徽蚌埠 233030)
金属有机骨架材料(MOFs)是由无机金属离子或金属簇和有机配体通过配位结合而形成的多孔材料,因金属离子的构型以及配体的配位方式不同可形成一维、二维和基于配位化学的三维结构[1]。MOFs 材料结合了无机化合物的刚性和有机聚合物的灵活性,具有独特的分子拓扑结构,具有孔隙率高、比表面积大、密度高、热稳定性好、结构多样以及通道尺寸可设计、可调整等特点,已被研究用于化学分离、气体储存、催化、非线性光学、药物输送和传感器技术等领域[2−6]。近年来,MOFs材料的研究也已经从简单的晶体结构研究逐渐演变为有目的的设计和基于功能特性合成目标晶体的研究。在含能材料研究领域,研究者们用含能配体或含能离子和金属构建了许多性能优良的含能金属有机骨架材料[7,8]。与传统含能材料叠氮化铅(LA)和苯乙烯酸铅(LS)等相比,这些材料具有能量密度高、稳定性好、灵敏度低、环境友好、高能配体可设计、爆轰性能可调节等优点,可被开发用于火炸药、燃烧催化剂、推进剂和烟火等领域[9,10]。高能MOFs结构的物理尺寸与灵敏度和性能密切相关,有序的三维框架结构有望具有理想的高密度、高能量输出、高热稳定性和低灵敏度,含能配体的结构和金属的选择均会对MOFs 的结构和最终的材料性能产生显著影响[11−13]。目前许多过渡金属,尤其是重金属如铅和镉离子常用于构建含能MOFs,但镧系金属的含能MOFs鲜有报道。
采用含羟基和羧基的配体3−硝基−4−(5−羟基−2,4−二硝基苯氧基)甲基苯甲酸,在水热条件下首次合成三种MOF:[Eu(HL)L(DMF)3]n(1),[Gd(HL)L(DMF)3]n(2)和[Tb(HL)L(DMF)3]n(3)。利用X射线单晶衍射、X射线粉末衍射、红外光谱和元素分析等方法对其晶体结构进行了分析,并对其热分解过程的非等温动力学进行了详细的讨论。
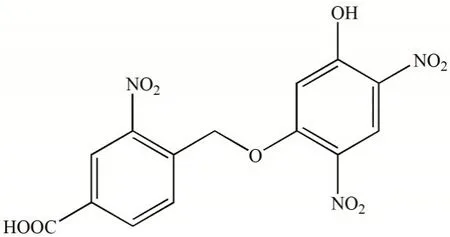
图1 H2L的结构式
一、实验部分
(一)试剂与仪器。仪器:Elementar Vario−EL CHNS 元素分析仪;NICOLET iS 50 傅立叶变换红外光谱仪;Bruker D8 ADVANCE X 射线单晶衍射仪;DX−2700BX−ray Diffractometer粉末衍射仪;STA 449−F5差示扫描量热仪。
试剂:实验中所用H2L 为济南恒化科技有限公司订制合成;Eu(NO3)3·6H2O分析纯,阿拉丁试剂(上海)有限公司;GdCl3·6H2O,分析纯,上海麦克林生化科技有限公司;Td(NO3)3·6H2O,分析纯,上海贤鼎生物科技有限公司;N,N′−二甲基甲酰胺(DMF),分析纯,西陇科学股份有限公司;无水乙醇,分析纯,成都市科隆化学品有限公司。
(二)配合物的制备。
1. 配合物[Eu(HL)L(DMF)3]n(1)的制备。将配体H2L(0.05mmol,17mg)和Eu(NO3)3·6H2O(0.05mmol,25mg)均匀分散在DMF−EtOH(3mL,1:2)混合溶剂中,加入玻璃样品管,于25mL反应釜中加热到80℃反应48h后自然冷却至室温,所得固体用DMF 反复洗涤后干燥,得到无色多面体形晶体。产率65%。IR:(KBr 压片,cm–1):3091(w),2930(m),2360(s),1658(m),1599(w),1535(s),1269(m),1105(w),831(w),748(w),447(w);元素分析(%),理论值(实测值):C 39.52(39.47);H3.14(3.11);N 11.22(11.19)。
2. 配合物[Gd(HL)L(DMF)3]n(2)的制备。将配体H2L(0.05mmol,17mg)和GdCl3·8H2O(0.1mmol,37mg)均匀分散在DMF−H2O(4mL,1:1)混合溶剂中,加入玻璃样品管,于25mL反应釜中加热到80℃反应48h后自然冷却至室温,过滤后得到棒状晶体。产率60%。IR(KBr 压片,cm–1):3426(w),3089(w),2473(w),1687(m),1538(w),1359(s),1246(w),877(m),672(w),443(w)。元素分析(%),理论值(实测值):C39.29(29.25);H3.12(3.09);N11.15(11.12)。
3. 配合物[Td(HL)L(DMF)3]n(3)的制备。将配体H2L(0.05mmol,17mg)和Td(NO3)3·6H2O(0.1mmol,45mg)均匀分散在DMF/H2O(3mL,2:1)混合溶剂中,加入玻璃样品管,于25mL 反应釜中加热至80°C 后反应48h,反应结束后自然冷却至室温,过滤后得到棒状晶体。产率62%。IR(KBr 压片,cm–1):3448(w),3068(w),2363(w),1655(s),1559(m),1312(s),1269(m),1061(w),831(w),748(w),675(w),443(w)。元素分析(%),理论值(实测值):C39.23(39.19);H3.11(3.09);N11.13(11.10)。
(三)配合物的结构测试。选取晶型较好、大小合适的配合物1的单晶,于Bruker Apex Smart APEX II型X射线单晶衍射仪上测定配合物的晶体结构,测定温度为172K,辐射源为Mo Ka(λ=0.71073Å),扫描方式为ω/2θ。共收集到7510 个衍射点,其中6489 个独立衍射点,将收集到的晶体数据分别用SAINT 和SADABS软件进行数据缩减和吸收校正。晶体结构用直接法解出(SHELXL−97),并用全距阵最小二乘法精修(SHELXL−97),氢原子位置按理论模型计算,并以统一的Uiso 值细化为骑乘原子[14]。配合物1的单晶衍射和结构精修参数见表1。由于配合物2 和3 的晶体数据不好,未得到符合精修要求的晶体结构。配合物2和3的晶胞参数与配合物1相似。配合物2的晶胞参数如下a=11.3795Å,b=29.1923Å,c=13.7147Å,β=105.1679,V=105.1679Å3。配合物3的晶胞参数如下a=11.3491Å,b=29.1404Å,c=13.7094Å,β=105.2322,V=4374.646Å3。X−射线粉末衍射证明配合物1−3 是同构的。配合物1 的CCDC 号码为2179535。

表1 配合物1的单晶衍射和结构精修参数
二、结果与讨论
(一)配合物的晶体结构。配合物1属于单斜晶系,Cc空间群,其结构及堆积图分别见图2−5,表2和表3分别列出了配合物1晶体结构的部分键长键角数据和氢键数据。如图2所示,配合物1的最小不对称单元含1个EuIII原子,1个羟基去质子化的配体HL−和1个羟基和羧基都去质子化的配体L2−,3个配位的DMF 分子。中心金属EuIII为9配位的多面体构型,分别与3个来自不同DMF分子中的O原子、2个来自配体中硝基的O原子、2个来自配体中羟基的O原子以及2个来自配体中的羧基的O 原子形成配位键。Eu-O 键长均在2.342(4)-2.795(5)Å 范围内。每个EuIII原子连接了4个配体,3个无序的DMF分子;每个配体的羧基和含有相邻羟基的硝基各连接了一个EuIII原子,即每个配体连接了2 个EuIII原子,成为直线性的2−连接构型,另外两个硝基没有与金属原子配位。整个结构是有4−连接的金属原子和2−连接的配体构成的三维网络骨架。在三维网络骨架中,一个配体中的两个苯环分别与另一个反向配体中的两个苯环接近平行,存在一强一弱的两组π−π共轭结构,其二面角分别为4.5(2)°和6.9(2)°,面心距分别为3.4973(2)Å 和3.9302(2)Å(图3)。除了苯环之间的π−π弱作用之外,配合物1的骨架中还存在分子内氢键(图4)。一个配体上羧基氢原子为质子给体,另一个配体上的去质子化的羧基氧原子为质子受体,形成了O−H…O分子间氢键。苯环上的氢原子为质子给体,另一个苯环上硝基氧原子为质子受体,形成了比较弱的C−H…O分子内氢键。由此可见,配合物1是一个含有π−π相互作用和分子内氢键的三维密堆积网络结构。沿着c轴方向存在椭圆形的孔道,孔道中充满了配位的DMF分子(图5)。

表2 配合物1的主要键长和键角数据
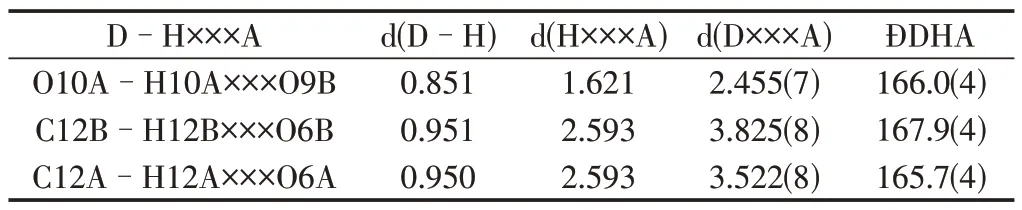
表3 配合物1的氢键数据

图2 化合物1的分子结构图(为清晰起见,对无序的DMF分子进行了简化)

图3 蓝绿色虚线为苯环之间的π-π相互作用
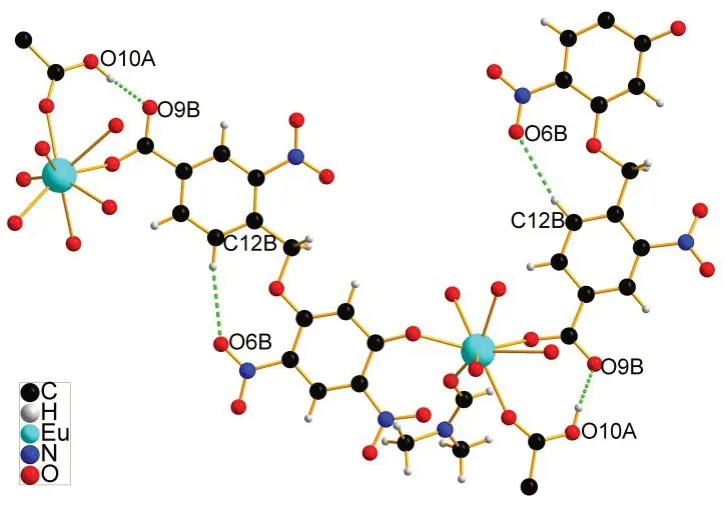
图4 绿色虚线为分子内氢键

图5 化合物1的3维结构(沿着a轴和c轴观看)
(二)配合物的X−射线粉末衍射和TG−DSC 分析。用粉末衍射仪确认了配合物1−3的纯度,配合物1测量的X−射线粉末衍射图谱主要衍射峰的位置与其根据其晶体结构模拟峰一致,证明配合物1样品是纯相。配合物2和3的X−射线粉末衍射峰和配合物1的晶体结构模拟峰一致,说明配合物2和3和配合物1是同构的,具体谱图见图6。

图6 配合物1-3的PXRD图谱
采用热重分析(TGA)和差示扫描量热法(DSC)研究了化合物配合物1−3的热分解行为。在N2流量为50mL/min,升温速率为5K•min–1,升温区间26−800℃的条件下,取配合物试样1.0mg左右进行测试,所得1−3的TGA−DSC曲线分别如图4a−c所示,配合物1−3的热分解曲线走向十分相近,也进一步说明各配合物分子的结构相似。
从图7 可以看出,配合物1 的DSC 曲线只有一个稍强烈的放热过程,峰值温度为261.8℃,对应TG曲线上的一个较快的质量损失阶段,从148.6℃开始,到330.2℃结束,失重比例为44.68%,之后均是缓慢的失重伴随着缓慢的放热过程,最终剩余质量分数为28.42%,推测最终产物可能是Eu2O3。固相分解过程的初始温度高于260℃,表明其具有良好的热稳定性。

图7 配合物1-3的TG-DSC曲线(5℃·min–1)
配合物2的TG曲线有两个质量损失阶段。第一个失重阶段发生在149.1~318.3℃,伴随着约51.87%的剧烈失重过程,应该是主骨架的坍塌,配体分解生成新的化合物,对应DSC曲线中放热峰值温度为261.7℃的明显的放热过程,第二个失重阶段发生在318.4~613.5℃,伴随着约46.23%的失重过程和DSC 曲线中放热峰值温度为537.8℃的剧烈放热过程,最终可能是由于样品爆炸离开坩埚而使坩埚中几乎没有残留样品。
配合物3经历了三个主要的连续质量损失阶段。第一个失重阶段为6.94%,从149.1℃开始,到177.0℃ 结束,损失的可能是结构中的溶剂分子,第二个阶段为183.8~344.6℃,质量损失为40.24%,对应DSC曲线中峰值为262.9℃的放热峰,此阶段应为配合物骨架分解阶段,随着温度继续升高,在344.0~670.3℃,配合物3经历了第三个失重阶段,失重36.5%,对应DSC曲线上一个巨大的放热峰,最终残留物的质量分数为16.55%,与Tb2O3的计算值(16.15%)一致,推测最终分解产物可能是Tb2O3。
(三)配合物热分解性能研究。为了讨论配合物1−3的热分解动力学,在5、10、15和20K·min–1四种不同升温速率(β)下,测得配合物的第一放热峰温度,具体数据列于表4,采用Kissinger 法和Ozawa−Doyle 法计算了配合物1−3的表观活化能(E)、指前因子(A)和线性相关系数(r)[15,16]。Kissinger(1)和Ozawa Doyle(2)方程分别如下:

表4 配合物1-3的放热峰温度和非等温反应动力学参数
式中,Tp为放热峰温度(K);β为升温速率(K·min–1);A为指前因子(s–1);R为气体常数(8.314J·mol–1·K–1);E为表观活化能(kJ·mol–1);C是一个常数。
从表4中可以明显看出,随着加热速率的升高,第一放热峰TP向更高的温度移动,线性相关系数非常接近1,通过两种方法计算所得到的活化能数值十分接近,这说明计算结果是可靠的。配合物的热分解温度较高,说明其热稳定性良好。配合物1−3 的Ea 值(Ek和E0的平均值)分别为171.850kJ·mol–1、137.555kJ·mol–1和149.500kJ·mol–1,说明三种配合物均具有良好的稳定性。通过以上计算结果,可将用于计算配合物1−3热分解过程的Arrhenius方程分别表示如下:
三、结语
以3−硝基−4−(5−羟基−2,4−二硝基苯氧基)甲基苯甲酸为起始原料合成了三种新型配合物[Eu(HL)L(DMF)3]n、[Gd(HL)L(DMF)3]n和[Tb(HL)L(DMF)3]n,并通过X射线单晶衍射、X射线粉末衍射、红外光谱和元素分析等方法对其结构进行了表征。配合物1 属于单斜晶系,Cc 空间群,中心金属原子EuIII与来自配体和DMF分子中的9个氧原子配位。每个EuIII原子连接了4个配体,每个配体连接了2个EuIII原子,形成了三维网络结构。此外,结构中具有π−π相互作用和分子内氢键,有助于提高其结构的稳定性。DSC 分析结果表明[Eu(HL)L(DMF)3]n分解是一个相对缓慢的放热过程,放热峰温为261.8℃、[Gd(HL)L(DMF)3]n和[Tb(HL)L(DMF)3]n的分解过程经历一个相对缓慢的放热过程和一个剧烈放热过程,第一放热峰温分别为261.7℃和262.9℃,采用Kissinger法和Ozawa−Doyle 法计算得到1−3的表观活化能分别为171.850kJ·mol–1、137.555kJ·mol–1和149.500kJ·mol–1。此研究可为含能稀土配合物的研究提供参考。
