深度学习在抗菌肽药物研究中的应用进展
2023-08-25朱尤卓刘红玉游宇豪郑珩
朱尤卓?刘红玉?游宇豪?郑珩

摘要:抗菌肽(AMP)作为未来最有希望解决病原微生物耐药性的新型抗菌药物之一,其研发备受关注。抗菌肽一般较短,组成多样,迄今人们已发现数千条天然抗菌肽,并建立了多个公开的抗菌肽数据库,为新型抗菌肽的研发和设计奠定了基础。另一方面,深度学习和人工智能作为信息处理的有力工具,已被大量应用于医学影像信息处理、疾病诊断、药物设计等领域,在抗菌肽的设计和研发上也受到广泛关注。在抗菌肽的信息描述方面,人们使用了伪氨基酸残基组成、位置特异性评分矩阵、独热码等多种特征向量;在深度学习方法上,研究人员应用了循环神经网络、卷积神经网络、对抗生成网络等多种算法,开发了ACEP、CLaSS等抗菌肽活性预测和序列生成模型。这些模型有望加速新型抗菌肽的发现,为应对耐药菌感染,尤其是臨床上难以治疗的耐药性革兰阴性菌感染,提供新的手段。
关键词:深度学习;抗菌肽;数据库;特征向量
中图分类号:R978.1文献标志码:A
Application progress of deep learning in antimicrobial peptide drug research
Zhu You-zhuo, Liu Hong-yu, You Yu-hao, and Zheng Heng
(School of Life Science and Technology, China Pharmaceutical University, Nanjing 211198)
Abstract As one of the most promising new antimicrobial therapy to solve the drug resistance of pathogenic microorganisms in the future, the research and development of antimicrobial peptides (AMP) has attracted much attention. Antimicrobial peptides generally have short sequences and diverse composition. By now, thousands of natural antimicrobial peptides have been discovered, and many public antimicrobial peptide databases have been established, which lays a foundation for the research and develop of new antimicrobial peptides. On the other hand, as powerful tools of information processing, deep learning and artificial intelligence have been widely used in medical image processing, disease diagnosis, drug design, and so on. They have also attracted extensive attention in the design and research of antimicrobial peptides. For the descriptors of antimicrobial peptides, people use a variety of feature vectors such as pseudo amino acid composition, position specific scoring matrix, and one-hot coding. In the deep learning method, researchers apply a variety of algorithms such as Recurrent Neural Network, convolutional neural network, and Generative Adversarial Networks, and develop the models of antimicrobial peptide activity prediction and sequence generation such as ACEP and CLaSS. These models are expected to accelerate the discovery of new antimicrobial peptides, and provide new means to deal with drug-resistant bacterial infection, especially drug-resistant Gram-negative bacterial infections that are difficult to treat clinically.
Key words Deep learning; Antimicrobial peptides; Database; Eigenvector
一份抗生素耐药性的评估报告指出,2050年可能有1000万人死于耐药细菌感染[1]。虽然目前上市的抗生素对治疗绝大多数感染仍有效[2],但由于人类长期广泛使用抗生素,越来越多的耐药菌出现,尤其令人担忧的多重耐药菌,包括屎肠球菌、金黄色葡萄球菌、肺炎克雷伯菌、鲍曼不动杆菌、铜绿假单胞菌、肠杆菌属等,它们造成了医院中的许多严重感染[3]。目前作为治疗耐药菌的最后手段的碳青霉烯类抗生素和黏菌素也开始面临耐药性的问题[4],因此需要新的抗菌药物来应对这一问题。
抗菌肽(antimicrobial peptide, AMP)是最有希望解决耐药菌问题的新型抗菌药物之一,序列一般较短,组成变化多样,但多为阳离子两亲性多肽分子,其抗菌作用具有多种可能机制,其中最常见的是通过与带负电荷的脂多糖(革兰阴性)或脂磷壁酸(革兰阳性)的磷酸基团的静电相互作用到达细胞膜,以库仑力吸附于细胞膜或进入细胞,随后膜破裂、细胞质渗漏,导致细菌死亡[5]。抗菌肽通过靶向整个细胞成分,而不是特定的分子,具有广谱的抗菌活性,同时避开了碳青霉烯类和替加环素等单一靶点药物的耐药性机制,该生化特性和药效学性质使其比传统抗生素更难耐药[6]。可惜的是,虽然迄今人们已发现成千上万条天然抗菌肽,且已有多个抗菌肽数据库被建立并公开,但是一方面由于抗生素新药研发耗时、昂贵、失败率高且盈利空间小,新抗菌药物的研发进入了冷门期,大型制药公司已基本放弃该市场[2],另一方面因抗菌肽结构不稳定性、多肽易降解和非特异性膜裂解的体内毒性等因素,限制了抗菌肽临床使用[7]。目前只有极少数公开的AMP获得美国食品和药物管理局(Food and Drug Administration, FDA)的批准[8]。
深度学习作为大数据处理的有力工具,已被大量应用于医学影像信息处理、疾病诊断、药物设计等领域,其有着高效且准确的判别能力,Stokes等[9]就利用深度神经网络从1.07亿个分子发现了在小鼠体内有广谱抗菌活性的新抗生素halicin,使用这一方法可在4 d内完成十多亿化合物分子的虚拟筛选,其效率远超传统的筛选手段。这是一种可以低成本、高效地发现活性高、毒性低以及结构稳定能临床应用的抗菌肽的新方法。
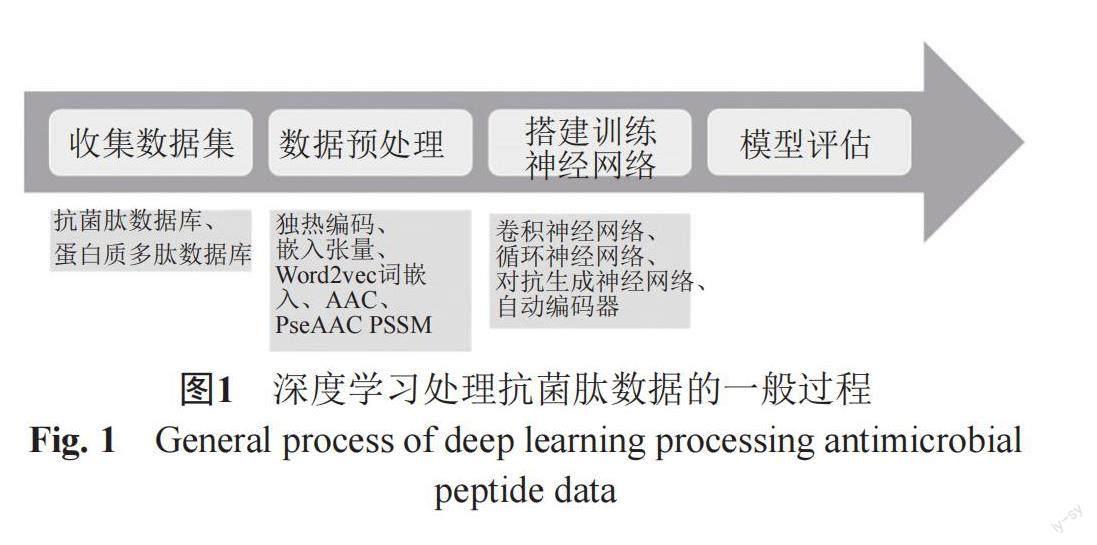
深度学习是根据经验(数据)自动确定深层网络参数的一门科学,它主要利用包含多个隐藏层的深层神经网络学习大量数据中的潜在规律以输出可靠结果,常见有监督和无监督学习两种方式。当通过训练而最小化的损失是网络输出和预先指定的期望输出(即训练集中的明确标签)之间的误差的度量时,训练被称为有监督的,如卷积神经网络(convolutional neural network, CNN)、循环神经网络(recurrent neural network, RNN)。当通过训练而最小化的损失不涉及一组样本输入的预先指定的期望输出时,该训练被称为无监督,如对抗生成网络(generative adversarial networks, GAN)、自动编码器(auto encoder, AE)[10]。深度学习方法虽然在大数据分析中具有普适性,但在不同应用中每个节点的处理上又具有特殊性,图1展示了深度学习处理抗菌肽数据的一般过程,前两步中数据来源与数据处理方法是明显区别于深度学习在其他方面的应用,另外,不同的神经网络模型具有不同的用途,CNN和RNN常被用于抗菌肽的活性预测[11-14],而GAN和AE则多被用于抗菌肽序列生成[15-17],但RNN有时也会被用于序列生成[18],这些都是值得我们特别去关注的。
1 收集数据集
抗菌肽又称宿主防御肽(host defensin peptide, HDP),广泛存在于自然界生物中[19]。自1922年發现溶菌酶开始,到1950年代左右发现杆菌肽和万古霉素,再到1980年代人们掀起一波对AMP的研究热潮,每年发现的AMP数量从1990年代的约50个增加到2000年的约100个[20],2010—2015年更是平均每年发表12,000篇相关文章[21],各种抗菌肽的序列、结构、活性以及修饰等信息越来越多被公开。研究者对这些信息进行收集整理,构建了许多抗菌肽数据库。表1展示一些通用抗菌肽数据库及其相关信息,更多特定的抗菌肽数据库可以查看文献[22]。
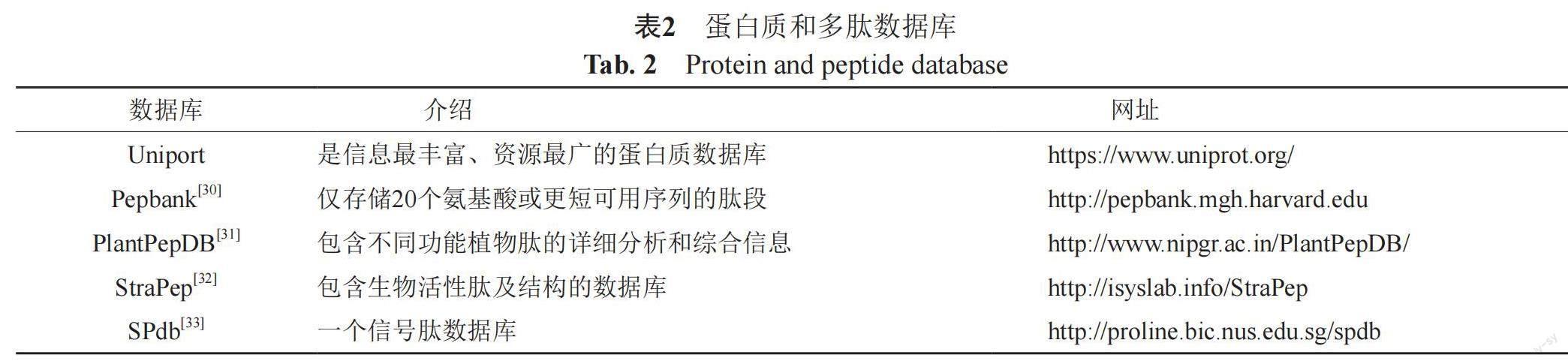
对于有监督学习,数据集中的每一个条目都有一个标签,作为期望输出。在抗菌肽活性预测等二分类问题中,这一标签通常是有活性(阳性)或无活性(阴性),具有不同标签的数据组成不同的数据集,阳性数据集常在抗菌肽数据库中收集,阴性数据集则多在其他蛋白质多肽数据库中收集,两者作为训练集和测试集输入神经网络。表2展示了一些常见的蛋白质多肽数据库,更多的多肽数据库可以查看文献[29]。而对于无监督学习,数据不需要标注,Das等[17]就设计了可以在UniProt数据库中报告的所有肽序列(可能无注释)上训练的无监督学习模型。
2 数据预处理
数据的特征是决定神经网络训练上限的关键因素。对于收集到的抗菌肽和其他多肽数据集,往往需要预处理把它转化为神经网络可识别的数据,即构建抗菌肽的特征参数或特征描述符。抗菌肽特征构建不仅借鉴计算机科学处理序列问题时使用的独热编码(one-hot encoder)、特征张量嵌入(feature tensor embedding)和Word2vec词嵌入等方式,同时还伴随生物信息学和计算生物学的进步产生更复杂和更具描述性的特征,这些特征不仅与理化性质有关,而且与微观层面的顺序结构以及进化信息等有关,如氨基酸组成(amino acid composition, AAC),伪氨基酸组成(pseudo amino acid composition, PseAAC),位置特异性评分矩阵(position-specific scoring matrix, PSSM)等。
独热编码是一种较为经典的多肽序列编码方式。它指的是分配20个输入单元来描述一个蛋白质残基,在二十维空间中,如用向量[1, 0, 0, 0…0, 0, 0]表示丙氨酸,[0, 0, 0…0, 0, 0, 1]表示缬氨酸[34]。独热编码作为一种多肽序列特征能在一定程度上反应多肽的序列信息,但它数据过于离散,很难捕捉到氨基酸之间的相似之处和不同之处[35]。特征张量嵌入则能较好地解决这一点,它利用概率生成的张量对氨基酸残基编码,该编码成为模型可训练的一部分,将氨基酸映射到可训练的实数张量,使用反向传播算法不断更新这些实数张量,氨基酸之间的相似性和差异性便可通过张量之间的几何距离来度量[11,35]。Word2vec词嵌入是自然语言处理中的一种网络模型,基于从大量文档语料库中收集邻近的单词数据,通过训练数据所学得的参数,即隐层的权重矩阵,生成该词语具有上下文属性的嵌入特征向量,其中类似向量往往分配给出现在类似上下文中的单词。Hamid等[36]把多肽序列中的连续3个氨基酸作为一个“词”,然后利用Word2vec中的skip-gram模型生成的词嵌入向量,用于细菌素识别。
氨基酸组成是Nakashima和Nishikawa在1994年提出的,它现在一般指多肽序列中20种氨基酸分别出现的频率,是一个有20个组分的向量[37-38]。在此基础上,发展出了伪氨基酸组成,其利用位置间隔为λ的氨基酸的疏水性值、亲水性值以及侧链质量等(都进行归一化处理)计算λ阶相关系数(θλ),若以向量X表示多肽的伪氨基酸组成,那么X中的前20个组分是归一化处理后第i种氨基酸出现频率?i(i=20),反映了氨基酸组成的影响,后λ个元素是归一化处理后有一定权重值ω的θλ,反映了氨基酸顺序和理化性质的影响[39]。现人们可以通过网页服务器http://chou.med.harvard.edu/bioinf/PseAA/生成所需的PseAAC[40]。在PseAAC的基础上,还发展出了伪K-tuple减少氨基酸组成(pseudo K-tuple reduced amino acids composition, PseKRAAC)[41]等方法。
位置特异性评分矩阵(PSSM)是进化信息的一种常见表示[42],一个长为L的多肽序列中,其每一个位置氨基酸突变为20种氨基酸的概率就构成了大小为L×20的PSSM矩阵[43]。PSSM矩阵可以通过PSI-BLAST程序获得,被Fu等[11]用于抗菌肽识别并获得了不错的结果。
3 深度学习模型及应用
深度学习是由多个处理层组成的计算模型,可学习具有多个抽象特征的数据,并通过反向传播算法来指示机器应该如何更新内部参数,从而发现大数据集中的复杂结构,它已经在预测潜在药物分子的活性等方面击败了其他机器学习技术[44],同时它在抗菌肽抗菌活性预测以及序列生成等方面也有着不错的表现。
卷积神经网络(CNN)的基本结构由输入层、卷积层(convolutional layer)、池化层(pooling layer,也称为取样层)、全连接层及输出层构成,可以有效地降低网络的复杂度,减少训练参数的数目,使模型对平移、扭曲、缩放具有一定程度的不变性,并具有强鲁棒性和容错能力,且也易于训练和优化[45]。Yan等[12]利用PseKRAAC和卷积神经网络开发了一个基于序列的短AMP分类模型,称为Deep-AmPEP30,该模型准确率比现有的基于机器学习的方法提高了77%,并且发现了与氨苄青霉素活性相当的抗菌肽P3(FWELWKFLKSLWSIFPRRRP)。
循环神经网络(RNN)是一类非常强大的用于处理和预测序列数据的神经网络模型,通过隐藏层上的回路连接,使得前一时刻的网络状态能够传递给当前时刻,当前时刻的状态也可以传递给下一个时刻[46],使得序列中的元素相互关联,另外人们通过在RNN单元中引入输入门,输出门和遗忘门,构建了长短期记忆模型(long short-term memory, LSTM),不仅提高标准循环单元的记忆能力,同时也解决了长期依赖的问题[47]。Wang等[18]搭建基于LSTM和双向LSTM的模型成功地生成并筛选到可能具有抗大肠埃希菌活性的新型AMPs。
深度学习用于抗菌肽研究时,往往不局限于用单一的神经网络分析抗菌肽数据集。Daniel等[13]就构建了一个包含嵌入层(embedding layer),卷积层(convolutional layer),最大池化层(max pooling layer)和LSTM层的深层神经网络模型,可以正确识别超过 98% APD 3数据库中的对革兰阳性或革兰阴性细菌具有活性的AMP。
另外深度生成模型也被用于抗菌肽序列的自动生成。自动编码器(AE)可通过编码器和解码器学习输入分子特征(及其属性),然后在潜在数据空间进行双向映射来生成新的分子,已被用于设计一个完全自动化的计算框架CLaSS。CLaSS使用自动编码器在多肽分子信息构建的潜在数据空间上进行训练,再利用线性插值的方法在空间中采样生成新的多肽序列,然后使用深度学习分类器以及从高通量分子动力学模拟得出的物理化学特征,来筛选生成的多肽分子,可用于广谱的AMP序列的从头设计与筛选。Das等[17]使用该方法获得两条对各种革兰阳性和革兰阴性病原菌(包括多重耐药的肺炎克雷伯菌)具有较高效力的抗菌肽YI12(YLRLIRYMAKMI)和FK13(FPLTWLKWWKWKK),同时它们在小鼠实验中也显示了较低的毒性。除了自动编码器,生成对抗网络(GAN)也被用于产生新的抗菌肽,它通过生成模型和判别模型的相互博弈学习,而产生较好的输出结果。Tucs等[15]设计的PepGAN模型可以控制生成序列的概率分布,使之尽可能多地覆盖活性肽,用该模型生成了一个最低抑菌浓度仅为氨苄西林一半的高活性抗菌肽AMP4 (GLKKLFSKIKIGSALKNLA) 。表3总结了一些用于抗菌肽研究的深度学习模型。
4 模型的评估及不足之处
对于深度学习模型的评估,通常包括计算和实验两大类的方法,在计算上常使用灵敏度(sensitivity)、特异性(specificity)、准确率(accuracy)以及馬修相关系数(matthews correlation coefficient,MCC)等作为评估指标,使用测试数据集来判断模型的准确性。但由于现有的抗菌肽特征表示方法,尚难以完整地描述抗菌肽特征,也缺乏可以模拟和描述AMP各种结构及物理化学特性的坚实理论[49],其生成和预测结果并不完全可信,因此常常需要与其他方法结合加以验证,比如Puentes等[50]提出了4种新兴技术相结合的抗菌肽设计筛选流程,包括人工智能、分子动力学、微生物表面展示(surface-display in microorganisms)和微流控(microfluidics),前两个是筛选和设计的计算机策略,而后两个对应于实验方法的合成和测试。使用实验合成并测试设计筛选的新型抗菌肽的活性,可以更准确地评估模型的效果,同时也可以发现一些有潜力的新抗菌肽。
另一方面,限制抗菌肽臨床应用的一个问题是毒副作用相对较大,尤其是溶血性问题,但是深度神经网络需要大量数据进行学习,目前可收集到抗菌肽相关溶血毒性实验数据较少,因此对于抗菌肽溶血毒性等深度学习预测模型也较少。在小分子药物毒性预测方面,人们已经开发了一些数据库和算法,如ToxAlert[51]和商业软件Discovery Stadio中的ADMET模块,这些方法通过统计或机器学习的方法,归纳出潜在的毒性结构基团,用于化合物分子的毒性预测。但是对于多肽,尤其是天然氨基酸组成的多肽,在其组成成分上通常没有明确的毒性基团,另一方面由于多肽结构的欠缺,也使得基于结构毒性基团预测较为困难。因此,在该领域还需要加强相关的研究,以促进抗菌肽的临床应用。
5 总结与展望
目前,深度学习和人工智能技术可以加速药物发现,在很大程度上为抗耐药菌感染药物的研发提供了新的化合物。对于只含天然氨基酸的抗菌肽序列,上述多种特征构建方法可用于深度学习,以预测抗菌活性或产生新的抗菌肽,但对于含复杂修饰且未知空间结构的抗菌肽,如订书肽(即在多肽结构中加入一个碳氢侧链或其他类型侧链以稳定其二级结构的多肽)[52],尚缺乏合适的结构表征方法,并且由于相应非天然抗菌肽的数据量较少,难以构建深度学习模型。可喜的是,深度迁移学习以及图神经网络等新的算法出现,有希望解决这些难题,前者可以在小数据集数据不足的情况下,先在大数据集上预训练,然后在特定目标数据集(即小数据集)上微调模型参数以实现模型在小数据集上的良好表现[53],后者则是能将多肽分子中原子和键转变为节点与边的图结构进行学习,实现对多肽复杂结构的表征,已被用于多肽毒性的预测[54]。同时抗菌肽等多肽以及蛋白质的数据库在不断地完善和丰富,更大的数据源变得公开可用,这些数据可以被进一步挖掘,并用于探索化学空间的新领域[55]。因此随着计算方法的发展和抗菌肽数据的增加,以深度学习为代表的人工智能方法,有望成为应对多重耐药菌问题以及发现新型抗菌药物的重要技术手段。
参 考 文 献
ONiel J. Tackling drug-resistant infections globally: Final report and recommendations[R]. London: Government of the United Kingdom, 2016: 1.
Rdal C, Balasegaram M, Laxminarayan R, et al. Antibiotic development-economic, regulatory and societal challenges[J]. Nat Rev Microbiol, 2019, 18(5): 267-274.
Tommasi R, Brown D G, Walkup G K, et al. ESKAPEing the labyrinth of antibacterial discovery[J]. Nat Rev Drug Discov, 2015, 14(8): 529.
沙国萌, 陈冠军, 王禄山. 抗生素耐药性的研究进展与控制策略[J]. 微生物学通报, 2020, 47(10): 3369-3379.
Lazzaro B P, Zasloff M, Rolff J. Antimicrobial peptides: Application informed by evolution[J]. Science, 2020, 368(6490): eaau5480.
Nagarajan D, Roy N, Kulkarni O, et al. Ω76: A designed antimicrobial peptide to combat carbapenem- and tigecycline-resistant Acinetobacter baumannii[J]. Sci Adv, 2019, 5(7): eaax1946.
Mourtada R, Herce H D, Yin D J, et al. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice[J]. Nat Biotechnol, 2019, 37(10): 1186-1197.
Annunziato G, Costantino G. Antimicrobial peptides (AMPs): A patent review (2015-2020)[J]. Expert Opin Ther Patents, 2020, 30(12): 931-947.
Stokes J M, Yang K, Swanson K, et al. A deep learning approach to antibiotic discovery[J]. Cell, 2020, 180(4): 688-702.e13.
Kriegeskorte N, Golan T. Neural network models and deep learning[J]. Curr Biol, 2019, 29(7): R225-R240.
Fu H, Cao Z, Li M, et al. ACEP: Improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding[J]. BMC Genomics, 2020, 21(1): 597.
Yan J, Bhadra P, Li A, et al. Deep-AmPEP30: Improve short antimicrobial peptides prediction with deep learning[J]. Mol Ther-Nucl Acids, 2020, 20: 882-894.
Daniel V, Uday K, Amarda S. Deep learning improves antimicrobial peptide recognition[J]. Bioinformatics, 2018, 34(16): 2740-2747.
Müller A T, Hiss J A, Schneider G. Recurrent neural network model for constructive peptide design[J]. J Chem Inf Model, 2018, 58(2): 472-479.
Tucs A, Tran D P, Yumoto A, et al. Generating ampicillin-level antimicrobial peptides with activity-aware generative adversarial networks[J]. ACS Omega, 2020, 5(36): 22847-22851.
Dean S N, Walper S A. Variational autoencoder for generation of antimicrobial peptides[J]. ACS Omega, 2020, 5(33): 20746-20754.
Das P, Sercu T, Wadhawan K, et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations[J]. Nat Biomed Eng, 2021, 5(6): 613-623.
Wang C, Garlick S, Zloh M. Deep learning for novel antimicrobial peptide design[J]. Biomolecules, 2021, 11(3): 471.
Boparai J K, Sharma P K. Mini review on antimicrobial peptides, sources, mechanism and recent applications[J]. Protein Pept Lett, 2020, 1(27): 4-16.
Wang G. The antimicrobial peptide database provides a platform for decoding the design principles of naturally occurring antimicrobial peptides[J]. Protein Sci, 2019, 29(1): 8-18.
Ageitos J M, Sánchez-Pérez A, Calo-Mata P, et al. Antimicrobial peptides (AMPs): Ancient compounds that represent novel weapons in the fight against bacteria[J]. Biochem Pharmacol, 2017, 133: 117-138.
Liu S, Fan L, Sun J, et al. Computational resources and tools for antimicrobial peptides[J]. J Pept Sci, 2017, 23(1): 4-12.
Wang G, Li X, Wang Z. APD3: The antimicrobial peptide database as a tool for research and education[J]. Nucleic Acids Res, 2016, 44(1): 1087-1093.
Hanif W F, Shankar B R, Pratima G, et al. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides[J]. Nucleic Acids Res, 2016, 44(1): 1094-1097.
Shi G B, Kang X Y, Dong, F Y, et al. DRAMP 3.0: An enhanced comprehensive data repository of antimicrobial peptides[J]. Nucleic Acids Res, 2021, 50(1): 488-496.
Malak P, Amstrong A A, Maia G, et al. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics[J]. Nucleic Acids Res, 2021, 49(1): 288-297.
Jhong J H,Chi Y H,Li W C, et al. dbAMP: An integrated resource for exploring antimicrobial peptides with functional activities and physicochemical properties on transcriptome and proteome data[J]. Nucleic Acids Res, 2018, 47(1): 285-297.
Ye G, Wu H, Huang J, et al. LAMP2: A major update of the database linking antimicrobial peptides[J]. Database, 2020, 2020: baaa061.
Dong F Y, Zhao G L, Tong H, et al. The prospect of bioactive peptide research: A review on databases and tools[J]. Curr Bioinform, 2020, 16(4): 494-504.
Duchrow T, Shtatland T, Guettler D, et al. Enhancing navigation in biomedical databases by community voting and database-driven text classification[J]. BMC Bioinformatics, 2009, 10(1): 317.
Das D, Jaiswal M, Khan F N, et al. PlantPepDB: A manually curated plant peptide database[J]. Sci Rep, 2020, 10(1): 2194.
Wang J, Yin T, Xiao X, et al. StraPep: a structure database of bioactive peptides[J]. Database, 2018, 2018: bay038.
Choo K H, Tan T W, Ranganathan S. SPdb-a signal peptide database[J]. BMC Bioinformatics, 2005, 6: 249.
Lin K, May A, Taylor W R. Amino acid encoding schemes from protein structure alignments: Multi-dimensional vectors to describe residue types[J]. J Theor Biol, 2002, 216(3): 361-365.
ElAbd H, Bromberg Y, Hoarfrost A. Amino acid encoding for deep learning applications[J]. BMC Bioinformatics, 2020, 21(10): 660-668.
Hamid M N, Friedberg I, Hancock J. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks[J]. Bioinformatics, 2018, 35(12): 2009-2016.
Nakashima H, Nishikawa K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies[J]. J Mol Bio, 1994, 238(1): 54.
Guo Z, Yang S, Hu Q, et al. A transverse and longitudinal encoding of protein sequence and its application[J]. J Comput Theor Nanosci, 2013, 10(2): 271-275.
Chou K C. Prediction of protein cellular attributes using pseudo-amino acid composition[J]. Proteins, 2001, 43(3): 246-255.
Shen H B, Chou K C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition[J]. Anal Biochem, 2008, 373(2): 386-388.
Zuo Y, Yuan L, Chen Y, et al. PseKRAAC: A flexible web server for generating pseudo K-tuple reduced amino acids composition[J]. Bioinformatics, 2016, 33(1): 122-124.
Liu Y, Gong W, Yang Z, et al. SNB-PSSM: A spatial neighbor-based PSSM used for protein-RNA binding site prediction[J]. J Mol Recognit, 2021, 34(6): e2887.
Ruan X, Zhou D, Nie R, et al. Predictions of apoptosis proteins by integrating different features based on improving pseudo-position-specific scoring matrix[J]. Biomed Res Int, 2020, 2020: 4071508.
Lecun Y, Bengio Y, Hinton G. Deep learning[J]. Nature, 2015, 521(7553): 436.
周飛燕, 金林鹏, 董军. 卷积神经网络研究综述[J]. 计算机学报, 2017, 40(6): 1229-1251.
杨丽, 吴雨茜, 王俊丽, 等. 循环神经网络研究综述[J]. 计算机应用, 2018, 38(S2): 1-6, 26.
Yu Y, Si X, Hu C, et al. A review of recurrent neural networks: LSTM cells and network architectures[J]. Neural Comput, 2019, 31(7): 1235-1270.
Dean S N, Alvarez J, Dan Z, et al. PepVAE: Variational autoencoder framework for antimicrobial peptide generation and activity prediction[J]. Front Microbiol, 2021, 12: 725727.
Torres M, Fuente-Nunez C. Reprogramming biological peptides to combat infectious diseases[J]. Chem Commun, 2019, 55(100): 15020-15032.
Puentes P R, Henao M C, Torres C E, et al. Design, screening, and testing of non-rational peptide libraries with antimicrobial activity: In silico and experimental approaches[J]. Antibiotics-Basel, 2020, 9(12): 854.
Sushko I, Salmina E, Potemkin V A, et al. ToxAlerts: A web server of structural alerts for toxic chemicals and compounds with potential adverse reactions[J]. J Chem Inf Model, 2012, 52(8): 2310-2316.
Tan Y S, Lane D P, Verma C S. Stapled peptide design: Principles and roles of computation[J], Drug Discov Today, 2016, 21(10): 1642-1653.
Cai C, Wang S, Xu Y, et al. Transfer learning for drug discovery[J]. J Med Chem, 2020, 63(16): 8683-8694.
Wei L, Ye X, Xue Y, et al. ATSE: A peptide toxicity predictor by exploiting structural and evolutionary information based on graph neural network and attention mechanism[J]. Brief Bioinform, 2021, 22(5): bbab041.
Melo M, Maasch J, De La Fuente-Nunez C. Accelerating antibiotic discovery through artificial intelligence[J]. Commun Biol, 2021, 4(1): 1050.
