生物信息学鉴定前列腺癌枢纽基因
2023-08-22张英郎张伟武燕龙包国昌
张英郎 张伟 武燕龙 包国昌
(赤峰学院附属医院泌尿外科赤峰市泌尿外科研究所,内蒙古 赤峰 024000)
前列腺癌(PC)是困扰全世界男性的主要癌症,好发于中老年人,国际癌症研究机构统计了2020年全球癌症发病率与死亡率,其中约有140万PC新发病例,37.5万人死亡。北欧,西欧,加勒比,澳大利亚/新西兰和北美位列发病率前5位〔1〕。我国由于人口老龄化加剧,生活方式及饮食结构的改变,其发病率也呈明显上升趋势。PC起病隐匿,临床发现时多伴有远处脏器及淋巴结,预后较差。生物信息学分析是基于组织细胞的芯片或高通量测序微阵列数据,将信息学与表达谱分析技术相结合,作为一个强有力的方法从成千上万个基因中挖掘有价值的基因,有助于解析癌症的潜在分子机制。GEO收录了世界各国研究机构提交的芯片或高通量表达数据,属于获得伦理批准的公共数据库。用户可以检索他人上传的一些实验测序数据并免费下载进行研究,所以不存在伦理问题和其他利益冲突。本研究采用此方法从中有效的筛选出了25个差异表达基因(DEGs),识别了4个与PC预后相关的筛选枢纽(Hub)基因,这些基因可能作为将来抗肿瘤药物开发及肿瘤发生发展机制研究的靶点,被进一步研究。
1 资料与方法
1.1数据获取与DEGs的鉴定 在GEO数据库中(https://www.ncbi.nlm.nih.gov)下载PC的mRNA微阵列芯片数据GSE46602〔2〕和GSE104749〔3〕,前者包含通过激光切除术获取的36例PC组织和14例正常组织,后者包含通过细针穿刺活检获取的4例PC组织和4例正常组织,检测平台均为GPL570 Affymetrix Human Genome U133 Plus 2.0 Array。应用Limma包对数据进行分析,设置筛选标准(P<0.01,|logFC|>2)后得到显著DEGs,以logFC为正值表示该基因在癌中高表达,为负值表示其在癌中低表达,通过网络工具Venn diagram(http://bioinformatics.psb.ugent.be/webtools/Venn/)获得两个数据集重叠的DEGs。
1.2蛋白互作(PPI)网络构建及hub基因的筛选 STRING 11.0 Version (http://string-db.org)是一个整合已知的和预测的蛋白质-蛋白质关联数据库,该数据库可应用于2 031个物种,包含960万种蛋白和1 380万条蛋白质之间的相互作用,有助于挖掘核心的调控基因。在Homo sapiens中查询25个DEGs。利用Cytoscape获得可视化的PPI网络图,选择马修斯相关系数(MCC)拓扑分析方法在PPI网络中对关键蛋白的进行预测得到degree前5位的Hub基因。
1.3差异基因本体化(GO)功能和京都基因与基因组百科全书(KEGG)通路富集分析 Metascape(http://metascape.org)是一个强大的基因功能注释分析工具,它能够去除功能冗余的富集通路,用简单明了的条形图显示出最主要的结果,富集的通路也能以网络形式呈现,更易于理解通路或生物学过程之间的关系。将DEGs及Hub基因输入到此网站,进行GO功能和KEGG通路富集分析。随后通过GeneCards网站(https://www.genecards.org/)对每一个Hub基因进行详细的GO和KEGG通路富集注释。
1.4Hub基因表达差异验证及预后分析 我们分别在GEO和肿瘤与癌症基因组图谱(TCGA,https://portal.gdc.cancer.gov) 数据库下载了两个芯片的series Matrix file(s)数据及TCGA_GTEX-PRAD TPM格式的RNAseq数据,应用3.6.3 R包进行统计学计算及可视化,分析hub基因在癌组织中对比正常组织的表达差异及与临床变量〔Gleason评分、前列腺特异性抗原(PSA)水平和淋巴结转移状态〕的相关性。为了鉴定与预后相关的基因,利用GEPIA(http://gepia.cancer-pku.cn)进行总生存(OS)和无病生存(DFS)分析,以P<0.05筛选与预后相关的基因,进一步通过R包统计分析Hub基因之间的相关性。
1.5Hub基因预测生存效能评价 下载TCGA_GTEX-PRAD队列中的表达数据,筛选出包含5年生存状态资料的患者79例,其中4例5年内因肿瘤原因死亡,75例5年时仍存活;建立索引获得患者Hub基因的表达水平数据,采用pROC R包和ggplot2 R包构建受试者工作特征(ROC)曲线,计算曲线下面积(AUC)预测5年疾病特异性生存与否的能力,以AUC>0.7表示具有一定的预测效能。
1.6统计学处理 采用SPSS22.0软件进行独立样本t检验、单因素方差分析、Kaplan-Meier生存分析、Log-rank检验。
2 结 果
2.1DEGs的识别 应用limma包分析,以P<0.05,|logFC|>1为标准,相比于正常组织,GSE4-6602队列共筛选了372个高表达及641个低表达的DEGs,GSE104749队列则有134个高表达及175个低表达的DEGs,见图1。进一步以|logFC|>2为筛选标准,GSE46602队列共筛选了159个下调及74个上调的DEGs,GSE104749队列有93个下调及82个上调的DEGs,Ven图获得两个数据集中9个共同上调和16个下调的DEGs。其中上调的基因为DLX1,RRM2,ITGBL1,ASPN,TRPM4,TOP2A,INHBA,BICD1,AURKA;下调的基因为NEFH,SLC14A1,TRIM29,LIN00844,VSNL1,CYP3A5,COL4A6,DSC3,ID4,AOX1,PDE8B,HOXD10,GATA3,FOXQ1,ANGPT1,ARMCX1。
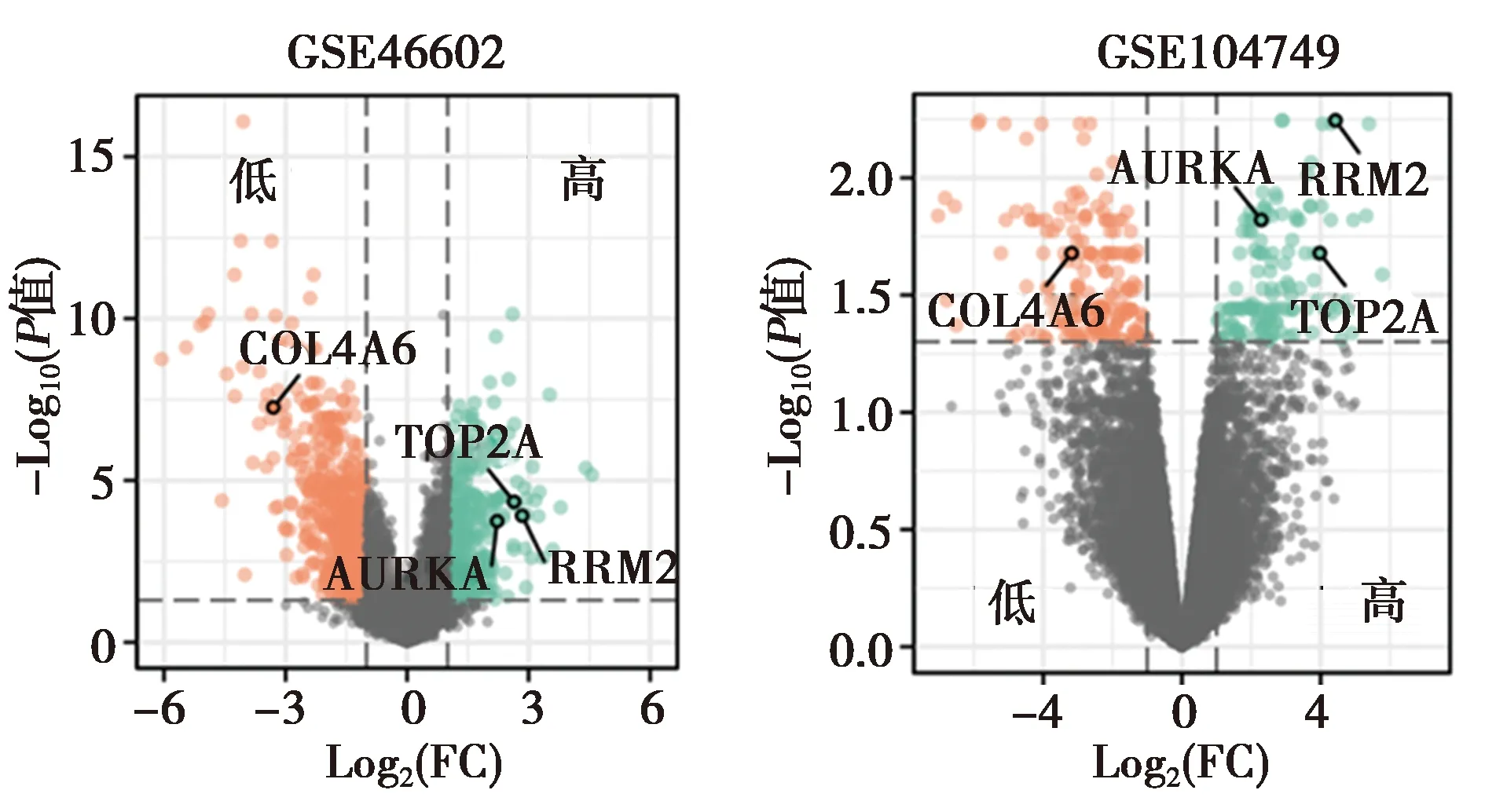
图1 差异表达基因火山图
2.2差异基因的PPI网络构建和Hub基因的筛选 将25个DEGs导入到STRING数据库中,得到了包含24个节点,9条互作关系线的PPI网络,PPI富集P<0.002 07,见图2。应用Cytohubba进一步筛选了排名前5位的Hub基因,分别为TOP2A,AURKA,RRM2,COL4A6,BICD1。

图2 25个DEGs的PPI网络
2.3差异基因的GO功能和KEGG通路富集分析 Metascape富集分析结果表明这25个DEGs共富集到7个GO生物学功能和1个KEGG信号通路上,主要集中神经元凋亡过程调控,激素水平调节,细胞周期负调控,药物代谢;5个Hub基因富集在细胞周期,有丝分裂通路中,见图3。GeneCards在线数据库对Hub基因分别进行详细的GO和KEGG注释,见表1。
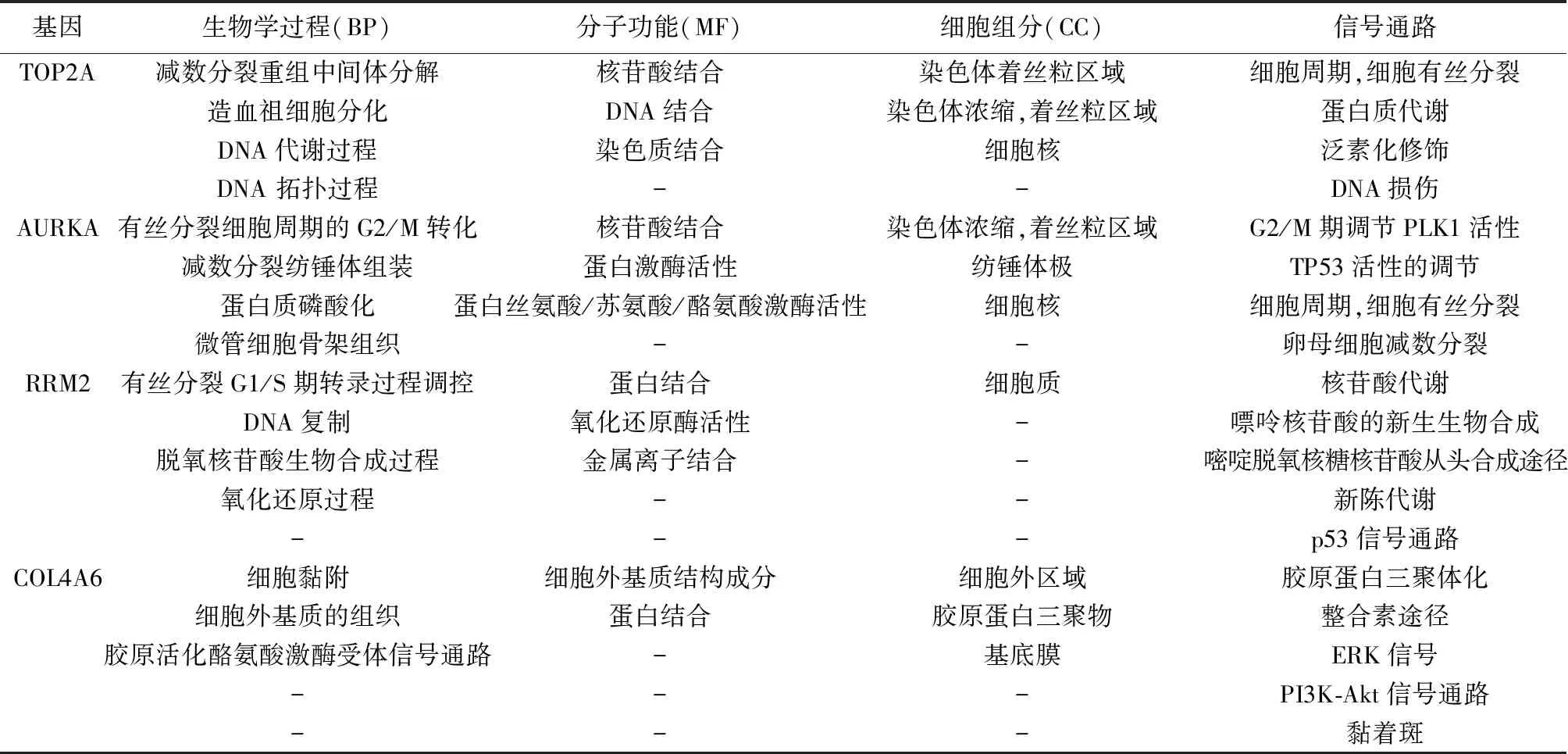
表1 GeneCards网站分析Hub基因主要参与的生物学功能和信号通路

图3 差异基因的GO和KEGG富集分析
2.4Hub基因在PC中的表达 基于GSE46602和GSE104749的芯片原始数据,将Hub基因的表达差异绘制成了散点图,见图4A。接着在TCGA_GTEX-PRAD大数据集中验证了这些基因在PC中的表达,结果显示,相比于正常前列腺组织,TOP2A,AURKA,RRM2及BICD1在PC组织中高表达,COL4A6在PC组织中低表达,见图4B~F,这与芯片结果相符。同时分析还表明TOP2A,AURKA及RRM2表达水平越高,其Gleason评分,PSA水平及淋巴结转移率越高(均P<0.05),COL4A6表达水平越低,Gleason评分,PSA水平及淋巴结转移越低(均P<0.05),见表2~4。

表2 Hub基因表达与PC患者Gleason评分的关系(M)

表3 Hub基因表达与PC患者PSA水平的关系(M)

表4 Hub基因表达与PC患者淋巴结转移的关系(M)
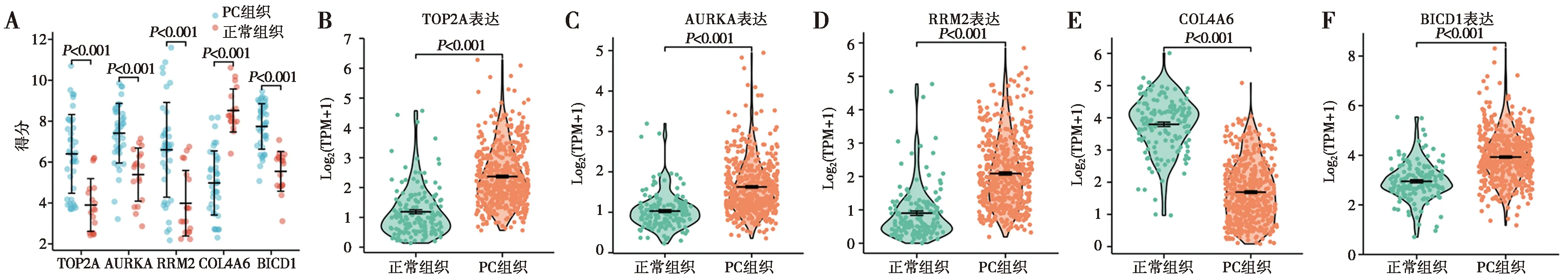
A:GSE46602和GSE104749数据集(PC组织n=40,正常组织n=18);B~F:TCGA_GTEx-PRAD数据集(PC组织n=496,正常组织n=152)
2.5Hub基因在PC中的预后分析及各基因的相关性分析 通过GEPIA网站对Hub进行生存分析,以中位数作为截断值分为高低表达两组,结果显示所有Hub基因在PC中的表达与患者OS无相关性(P>0.05),见图5,但与患者DFS显著相关。TOP2A,AURKA及RRM2在PC中高表达与PC患者更短的DFS显著相关(P<0.05),COL4A6在癌中低表达与较短的DFS显著相关(P<0.05),见图6。随后通过R在TCGA-PRAD-RNAseq数据集中对TOP2A,AURKA及RRM2的表达数据进行Shapiro-Wilk正态性检验,结果不满足正态分布(P<0.05)。随后用Spearman秩相关进行相关分析,结果显示TOP2A与AURKA及RRM2之间呈显著正相关(r=0.890、0.890,均P<0.001),RRM2与AURKA之间呈显著正相关(r=0.850,P<0.001)。

图5 Hub基因与OS的关系生存
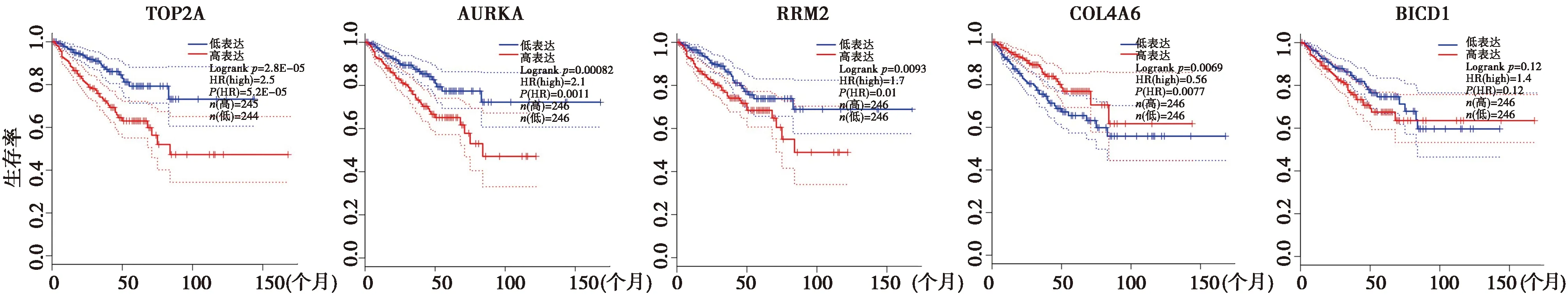
图6 Hub基因与DFS的关系生存
2.6Hub基因在PC患者5年疾病特异性生存与否中的预测效能 在TCGA_GTEx-PRAD队列中,TOP2A(AUC=0.754),AURKA(AUC=0.765),RRM2(AUC=0.826)及COL4A6(AUC=0.901)均表明在预测PC患者5年后是否生存有一定的准确性,其中RRM2表现最优,见图7。

图7 Hub基因预测PC患者5年疾病特异性生存的ROC曲线
3 讨 论
基因表达数据反映的是直接或间接测量得到的基因转录产物mRNA在组织细胞中的丰度,生物信息学可以利用这些数据分析哪些基因的表达发生了改变,基因之间有何相关性,在不同条件下基因的活动是如何受影响的,这在揭示疾病内在机制、发现治疗靶点、辅助诊断、药物疗效判断、预测疾病预后等方面有重要的作用。本研究的主要思路基于“挑”筛选DEGs、“圈”功能聚类、“联”蛋白互作、“靠”临床意义这四个维度,识别了4个具有临床意义的Hub基因。分析发现TOP2A,AURKA,RRM2在PC中高表达,而COL4A6在PC中表达降低,且这些基因的表达与PC患者Gleason评分,PSA水平及淋巴结转移具有显著相关性,提示它们与PC的侵袭能力及恶性程度相关,这对于肿瘤发生发展机制研究及后续药物靶点的研发有一定的参考价值。生存分析显示TOP2A,AURKA,RRM2作为致病性基因,其高表达与PC患者较短的DFS显著相关,而COL4A6作为保护性基因,其高表达与PC患者较长的DFS显著相关。而这些基因表达与OS无统计学差异,可能因前列腺癌预后良好,OS作为研究终点不易获得有关。表达谱分析中,经常会用到相关性分析,探索一组基因间的共表达特征。如这些基因间的表达是否存在较强的协同性,一个基因表达值的改变是否与另一个基因表达值改变显著相关,它们之间是共激活还是抑制关系等。本研究结果表明,TOP2A,AURKA及RRM2之间在PC中存在显著正相关,提示他们可能在相似的生物学过程中发挥作用。ROC曲线揭示这些基因在预测PC患者5年疾病特异性存活状态中均具有较高的敏感度和特异度。GO和KEGG富集分析进一步揭示它们共同参与细胞周期,有丝分裂,为深入研究分子机制提供了方向。
TOP2A全名脱氧核糖核酸拓扑异构酶(DNA Topoisomerase Ⅱ Alpha),编码基因定位于17q21.2,主要通过参与DNA分裂、修复、重组、复制、转录及染色体分离浓缩等过程调节DNA拓扑结构〔4〕。TOP2A通过调控DNA链的断裂和重新连接,从而影响DNA的拓扑状态和复制〔5〕,这种机制在肿瘤的增殖、转移和化疗药物耐药性等方面发挥重要作用〔6〕。TOP2A在人类大部分肿瘤中高表达,诱导癌细胞的持续增殖,并导致转移,影响患者的生存预后。Del Moral-Hernández等〔7〕利用免疫组化分析了1 485例宫颈刮片及活组织切片的细胞学样本,发现TOP2A/MCM2表达随宫颈上皮内麟状病变程度而升高,其高灵敏度和特异度是识别并预测宫颈癌前病变进展风险的最佳生物标志物,可普及临床应用于女性宫颈病变筛查中。Du等〔8〕通过生信分析并结合实验发现TOP2A在人肺腺癌(LUAD)中高表达并与患者OS预后相关,机制上通过细胞外调节蛋白激酶(ERK)/C-Jun氨基末端激酶(JNK)/p-P38/C/EBP同源蛋白(CHOP)信号通路促进肿瘤的进展,TOP2A可能会成为LUAD患者的预后标志物和潜在治疗靶点。Ren等〔9〕研究显示,TOP2A蛋白在不同亚型乳腺癌中均有显著表达,其在高增殖亚型乳腺癌〔如基底样、Luminal B、人表皮生长因子受体(HER)2阳性〕中表达更高,TOP2A高表达的乳腺癌患者预后较差。Won等〔10〕提出TOP2A可作为腋窝淋巴结阳性乳腺癌的预测标志物。Resende等〔11〕报道,TOP2A蛋白表达与较高的Gleason评分和术前PSA水平相关,且TOP2A水平较高的患者无生化复发生存期(BRFS)较短,在多因素Cox回归分析中,TOP2A仍然是BRFS的独立预后因素,这与本研究结果一致。此外以TOP2A为靶点的靶向药物研究也已应用于临床〔12,13〕,Jain等〔14〕在mRNA及蛋白层面同时验证了TOP2A在肾上腺皮质癌(ACC)中高表达,细胞实验表明TOP2A抑制剂阿柔比星具有最显著的抗癌细胞活性,阿柔比星可能是未来临床试验中用于局部晚期和转移性ACC患者有效的候选靶向药。Liu等〔15〕实验表明,TOP2A抑制剂氯化两面针碱可能成为治疗肝细胞癌的直接靶点。一项最新研究〔16〕显示,TOP2A抑制剂依托泊苷(VP-16)在CRPC模型中是有效的,侵袭性变异性PC患者的特定亚群可以从VP-16治疗中获益,TOP2A也是预测VP-16反应的良好生物标志物,雄激素受体(AR)信号通路也和VP-16之间存在紧密的联系,需要进一步深入研究。
AURKA是一种丝氨酸/苏氨酸激酶,编码基因定位于20q13.2,在细胞有丝分裂过程中发挥重要调控作用。AURKA作为一种癌基因,已经被发现在多种类型的癌症中发挥致癌基因的作用,包括实体肿瘤和恶性血液肿瘤,并可作为有效的治疗靶点〔17,18〕。基因扩增、转录激活和蛋白降解抑制可促进癌组织中AURKA表达的升高。AURKA通过参与癌细胞增殖、上皮-间充质转化(EMT)、凋亡和癌症干细胞的自我更新促进肿瘤发生。AURKA已被证实可调控多种癌症相关的信号通路,包括磷脂酰肌醇3激酶/蛋白激酶B(PI3K/Akt)、哺乳动物雷帕霉素靶蛋白(mTOR)、β-catenin/Wnt和核因子(NF)-κB通路,肿瘤发生需要多种信号通路之间的相互作用,提示AURKA在这些过程和通路中的重要意义〔19〕。一些高选择性的AURKA小分子抑制剂如MLN8273已被证明通过抑制有丝分裂、诱导细胞周期阻滞和自噬、加速癌细胞凋亡和衰老来抑制细胞增殖,目前正处于Ⅰ/Ⅱ/Ⅲ期临床实验中〔20~22〕。MLN8237能显著增加肿瘤细胞对化疗药物或放疗的敏感性〔23,24〕。新型内分泌治疗药物阿比龙或蒽杂鲁安问世能够直接靶向AR治疗PC,然而,接受AR定向治疗的PC人群可能进展为晚期CRPC,最终发展为神经内分泌前列腺癌(NEPC),这些患者丧失AR信号依赖或AR表达而对治疗产生耐药性,导致治疗选择受限。Ton等〔25〕通过深入的研究,设计了一种能同时抑制N-Myc和AURKA靶点的化合物,通过实验证明可以有效的作为神经内分泌PC的潜在治疗药物,但需要更广泛的优化及临床实验。由此可见以AURKA为靶点的抗癌药物研发拥有巨大的潜力。
RRM2又名核糖核苷酸还原酶调节亚基M2,编码基因定位于2p25.1,是DNA合成和修复的关键酶,RRM2同样参与了多种癌症的进展。Ma等〔26〕通过生信分析发现,无论在TCGA还是其他队列中,高RRM2表达是LUAD OS期、疾病特异性生存期和无进展生存期的独立危险因素,且RRM2与B细胞、CD8+及CD4+T细胞等免疫细胞浸润也显著相关,为未来研究提供更多的可能性和方向。Wang等〔27〕研究表明,在口腔鳞状细胞癌(OSCC)中,RRM2表达明显升高,过表达RRM2能够促进OSCC细胞的增殖和迁移,抑制细胞凋亡。且其高表达与更晚的病理分级和OSCC复发密切相关,RRM2高表达患者的OS率较低表达短。Lee等〔28〕等指出,RRM2可作为肝细胞癌根治性切除术后监测早期复发的敏感指标。Zhang等〔29〕通过PCR的方法检测了21例腹膜后脂肪肉瘤(RLPS)和10例正常对照组织,发现RRM2在癌组织中高表达,细胞实验表明沉默RRM2可经Akt/mTOR/4EBP1通路抑制细胞增、迁移和侵袭,促进细胞凋亡。体外裸鼠实验,RRM2抑制剂能够缩小肿瘤体积,有望成为治疗RLPS的有效靶点。Mazzu等〔30〕报道了RRM2能够通过Akt/mTOR,信号传导及转录激活蛋白(STAT),P53,DNA损伤修复,细胞周期,细胞凋亡等多种信号通路在PC中发挥致癌作用,并能导致EMT过程促进肿瘤侵袭及转移。另外RRM2过表达导致基因组不稳定性增加〔31〕,通过基因组改变的比例(FGA)测量的拷贝数变异已被证明与Gleason评分和PC患者转移的发展有关〔32〕,Taylor等〔33〕队列中也观察到RRM2水平与Gleason评分呈显著正相关,这也证实了本研究结论。
Ⅳ型胶原是存在于血管周围基底膜和皮肤真皮表皮连接处的主要胶原,由内皮细胞和周细胞合成,有助于血管的稳态和重构,包含个胶原链(α1至α6)〔34〕。基底膜在抑制肿瘤中起着至关重要的作用,当基底膜受损时,可促进癌症进展和转移〔35〕。COL4A6又名Ⅳ型胶原α6,编码基因定位于Xq22.3。研究〔36〕显示,COL4A6在PC中呈显著低表达及其启动子高甲基化,其表达下调可激活p-FAK/MMP-9信号通路促进肿瘤细胞的进展和转移,然而还需体外动物实验进一步验证及大规模PC患者队列评价。
综上,当前针对这些靶点的靶向药物研究正逐步被探索,以期挽救难治性及特殊亚型的PC患者,但其有效性还需开展大规模、开放性、多中心的临床实验得以推广应用。总之,生物信息学方法对数据库高效能的分析能力使得挖掘的结果具有极大参考意义,该方法有助于人类从基因组学、转录组学、蛋白组学、免疫组学、代谢组学、修饰组学及药理组学等多组学全面洞悉疾病机制,攻克疾病,可以合理利用现有信息资源,缩短科研周期,降低科研成本。
