猪圆环病毒3型与宿主相互作用机制研究进展
2023-08-22孙仁杰安慧婷王雅婷谢荣辉张传亮赵灵燕方维焕李肖梁
孙仁杰,单 颖,安慧婷,王雅婷,谢荣辉,张传亮,赵灵燕,方维焕,李肖梁,*
(1.浙江省动物疫病预防控制中心,浙江 杭州 311199; 2.浙江省动物预防医学重点实验室,浙江 杭州 310058; 3.浙江大学 动物科学学院,浙江 杭州 310058)
猪圆环病毒3型(porcine circovirus type 3,PCV3)属于圆环病毒科(Circoviridae)圆环病毒属(Circovirus),于2015年首次在美国母猪群中通过宏基因组测序技术发现,感染该病毒的母猪表现出繁殖障碍,以及与猪皮炎肾炎综合征(PDNS)相似的症状[1]。作为新发猪病病原,根据大量的流行病学回溯性调查研究,目前PCV3已在全球多个国家和地区的养殖业猪群和野猪群被检测到,并且在其他健康动物中也偶有发现[2-4]。目前发现的与PCV3感染相关的临床症状包括PDNS、繁殖障碍、呼吸系统紊乱、腹泻和系统性炎症反应综合征[2,5]。
与猪圆环病毒2型(porcine circovirus type 2,PCV2)相同,PCV3也是编码复制相关蛋白的环状单链DNA病毒(circular replication-associated protein-encoding single-stranded DNA viruses, CRESS DNA viruses)类群的一员[6]。PCV3基因组为2.0 kb,序列分析预测其至少包含3个开放阅读框(open reading frame,ORF)。最大的开放阅读框ORF1位于基因组正义链,沿顺时针方向转录,编码复制酶蛋白(Rep)。核衣壳蛋白(capsid protein,Cap)由位于基因组互补链的ORF2编码,转录方向与ORF1相对。目前ORF3的起始密码子和功能尚不清楚。此外,PCV3基因组全序列和Cap氨基酸序列与PCV1、PCV2相比同源性较低[1-2]。
受限于基因组大小和自身编码能力,PCV的复制和增殖在很大程度上依赖于宿主细胞,易引起宿主基因表达的异常和细胞功能形态的改变。因此,研究病毒与宿主之间的相互作用机制,并鉴定出参与的宿主因子和病毒组分,有助于明晰猪圆环病毒复制与发病机制。自PCV3发现以来,有关流行病学调查、检测方法建立和基因组演化分析的研究较多。部分实验室通过瞬转表达系统、临床分离和反向遗传学技术在病毒组分或全病毒水平对PCV3感染致病与宿主响应展开了研究。本文拟通过对PCV3感染引起宿主响应、与PCV3互作宿主因子鉴定和PCV3生命周期等相关的最新研究结果进行综述,并对相关作用机制进行探讨,有助于进一步了解PCV3复制及其感染致病机制。
1 PCV3感染与宿主响应
1.1 PCV3反向遗传学技术的应用研究
已在患病仔猪或者健康动物的大脑、心脏、脾、肾脏、口腔、血清、鼻腔液、胸腔积液、腹腔液、粪便和精液等大部分组织和体液中检测到PCV3[3,5,7-8]。大部分研究集中于PCV3的流行病学调查和检测方法上,缺乏活病毒株来研究其在猪源细胞甚至宿主体内的发病机制。Jiang等[7]将合成的PCV3线性基因组通过EcoR I和SacI酶切位点与pSK载体连接构建感染性克隆,转染猪肾上皮细胞(PK-15细胞),连续传15代,首次在体外拯救获得了PCV3毒株。之后,陆续有研究人员通过在PCV3基因组的酶切位点设计引物进行PCR获得线性DNA,利用T4连接酶将基因组在体外环化转染PK-15细胞或可传代猪肺泡巨噬细胞(3D4/21细胞),连续传代获得病毒[9-10]。一些科研者尝试使用多种载体和不同PCV3基因组序列构建感染性克隆质粒,经磷酸钙或脂质体转染不同细胞系或小鼠体内拯救均未成功获得PCV3拯救病毒[11-12]。PCV3拯救病毒在细胞或小鼠体内的传代增殖能力较弱,说明病毒增殖传代方法需要进一步研究优化,这也是目前有关PCV3致病机制研究较少的原因之一。
1.2 PCV3感染差异蛋白质组学分析
Jiang等[7]通过PCV3感染SPF仔猪的动物试验发现,病毒感染后可在仔猪肺部组织中观察到严重的病理变化。为进一步了解PCV3与宿主之间的相互作用及其发病机制,Jiang等[13]利用iTRAQ同位素标记定量结合LC-MS/MS质谱分析,对感染PCV3的无特定病原体仔猪肺部组织的细胞调控蛋白进行差异分析,共检测到3 429个蛋白,从感染组和对照组的比较分析中筛选到242个差异蛋白,其中上调蛋白100个,下调蛋白142个;差异蛋白质主要涉及细胞代谢、先天免疫反应、主要组织相容性复合体、热休克蛋白家族和细胞胞吞作用。
1.3 PCV3感染诱导细胞应激反应与相关适应性机制
在感染病毒的细胞中,病毒基因组复制与蛋白质的合成能引起细胞发生应激反应,从而在感染过程中改变细胞内环境。为应对病毒感染引起的外源性刺激,宿主细胞演化出多种适应性应答机制,包括未折叠蛋白反应、凋亡、自噬和线粒体自噬,以恢复细胞内环境稳态。
PCV2感染细胞通过激活AMPK/ERK/mTOR通路诱导细胞自噬,且自噬促进病毒复制[14-15]。Gu等[16]进一步分析发现,PCV2感染通过上调细胞内Ca2+含量,引起胞内Ca2+稳态失衡,继而激活CaMKKβ/AMPK/mTOR和CaMKKβ/CaMKI/WIPI1通路诱导细胞自噬,Cap是介导这一过程的主要病毒蛋白。那么,PCV3 Cap是否与PCV2 Cap具有相似的功能特性,诱导细胞自噬?Geng等[17]通过质粒转染和慢病毒介导的基因转导在HEK-293T细胞中高效表达PCV3 Cap蛋白以研究其诱导细胞自噬的机制,结果表明,PCV3 Cap能通过抑制哺乳动物雷帕霉素靶蛋白(mTOR)磷酸化从而诱导细胞发生自噬,并且泛素-蛋白酶体途径也参与这一过程。然而,在全病毒水平和宿主猪源细胞上PCV3是否能诱导细胞自噬还有待进一步研究。
细胞凋亡在病毒与细胞的博弈中扮演了重要角色。在感染早期,病毒通过抑制细胞凋亡来促进自身复制增殖;感染后期,病毒诱导细胞凋亡并促进子代病毒的释放和传播。因此,在病毒诱导细胞凋亡过程中病毒蛋白的抗细胞凋亡和诱导细胞凋亡活性非常重要[18]。与PCV2相似,PCV3 Cap能通过激活Caspase-9依赖的线粒体途径诱导PK-15细胞发生凋亡。并且PCV3 Cap的表达能将细胞周期阻滞于S期,从而抑制PK-15细胞的增殖[19]。
Jiang等[13]对仔猪感染PCV3的肺部组织进行了蛋白质组学分析,筛选到了超氧化物歧化酶2(SOD2),并通过免疫印迹和免疫组化进一步证实了其在PCV3感染后表达水平上调。Chen等[20-21]研究发现,PCV2感染诱导细胞氧化应激,可通过抑制细胞内谷胱甘肽氧化酶1(GPX1)的活性引起细胞内产生过量活性氧(ROS)。Sun等[22]研究发现,PCV2感染诱导的内质网应激和氧化应激之间存在密切联系,PCV2感染选择性激活PERK通路,诱导内质网氧化还原酶1α(endoplasmic reticulum oxidoreductase 1α,ERO1α)表达上调,引起内质网ROS的产生,进而引发氧化应激。因此,PCV3感染后SOD2表达水平上升可能是感染细胞内氧化还原状态失衡所致,即PCV3感染可能诱导氧化应激。
1.4 PCV3感染对宿主细胞先天免疫与炎症反应的影响
病毒在与宿主的博弈中演化出了一系列策略来削弱或逃逸细胞的抗病毒免疫,从而得以生存并感染增殖。PCV2已被证明是具有免疫抑制作用的病原微生物,能通过与宿主蛋白之间复杂的相互作用调控宿主免疫和炎症反应[23]。与PCV2相似,临床感染PCV3的猪个体会出现多器官炎症,且存在与其他致病性病原共感染的情况,这可能与PCV3感染引起宿主免疫炎症反应紊乱相关。同时,有研究报道,在健康猪群中也存在PCV3感染,提示PCV3能逃逸宿主抗病毒免疫[2]。
宿主先天免疫反应是细胞通过模式识别受体(PRR)识别病原体携带的病原体相关分子模式(PAMP),并激活、介导下游相关免疫信号通路,进而诱导释放细胞因子和干扰素。在HEK-293T细胞中的研究结果显示,PCV3 Cap能激活NF-κB信号通路和诱导促炎细胞因子IL-6和TNF-α的转录表达。并且PCV3 Cap能上调RIG-I样受体(RLRs)信号通路中的RIG-I/MDA5和Toll样受体(RLRs)信号通路中的MyD88的转录水平[24]。但PCV3 Cap诱导激活NF-κB信号通路的具体机制有待进一步探索。在3D4/21细胞中瞬时转染表达PCV3 Rep和Cap能影响促炎细胞因子TNF-α、IL-6和免疫抑制细胞因子IL-10的mRNA转录。这与前期研究报道仔猪接种PCV3拯救病毒的体内实验中,血清IL-10水平持续上升的结果一致,提示PCV3感染可能通过诱导IL-10上调使宿主产生免疫抑制[7]。李萌[25]初步研究了PCV3相关蛋白诱导3D4/21细胞IL-10表达的分子机制,结果表明,PCV3 Cap相较于Rep可能是诱导IL-10上调表达的主要蛋白;与PCV2 Cap相似,NF-κB信号通路参与PCV3 Cap诱导IL-10表达上调的过程[25]。目前有关PCV3感染对细胞因子表达和炎症反应影响的研究较少,后续相关探索将有助于更好理解PCV3亚临床感染向临床疾病发展的进程,以及与其他病原并发感染的可能性。
干扰素(IFN)具有广谱抗病毒特性,是宿主先天免疫反应中重要的细胞因子,在宿主抵御病毒感染的过程中发挥重要作用。PCV2感染细胞能通过相关信号通路诱导IFN-α和IFN-γ表达,且二者能促进PCV2复制[23]。在3D4/21细胞中的研究发现,PCV3 Rep和Cap均能上调IFN-β和IFN-γ的表达,而在诱导IFN-α表达上,Cap和Rep呈现了相反的效应[25]。Jiang等[13]通过蛋白质组学分析发现,在PCV3感染的仔猪中,2′,5′-寡腺苷酸合成酶(OAS1)、干扰素诱导的GTP结合蛋白(Mx1和Mx2)、人干扰素刺激基因表达蛋白15(ISG15)、干扰素诱导的三角形四肽重复蛋白3(IFIT3)和干扰素诱导蛋白44(IFI44)等干扰素刺激基因(ISGs)表达蛋白显著上调,提示这些ISGs蛋白可能参与PCV3感染诱导的宿主先天免疫应答。申翰钦[26]和Zhang等[27]围绕PCV3 Cap对外源性DNA诱导细胞产生IFN-β的影响展开研究,结果显示,PCV3 Cap抑制由刺激IFN表达DNA(ISD)诱导的IFN-β mRNA转录、IFN启动子激活和TANK结合激酶1(TBK1)磷酸化,但不影响环鸟苷酸-腺苷酸合成酶(cGAS)/干扰素基因刺激蛋白(STING)诱导的IFN启动子激活,推测PCV3 Cap可能通过干扰cGAS与ISD的结合从而抑制IFN产生。利用蛋白质体外结合实验、LC/MS及免疫共沉淀技术,研究人员发现PCV3 Cap能竞争性与G3BP1结合从而抑制cGAS与G3BP1的相互作用,并且证明G3BP1参与DNA诱导的IFN-β产生且存在正向调控作用。因此,PCV3 Cap 通过与G3BP1互作干扰cGAS识别ISD从而抑制IFN表达。申翰钦[26]和Shen等[28]进一步研究PCV3 Cap对IFN-β诱导ISG表达的影响与相关机制,结果显示,PCV3 Cap抑制IFN-β诱导的IFIT1和IFIT2转录,以及干扰素刺激应答元件(ISRE)启动子的激活,提示其抑制IFN-β激活的两面神激酶/信号传导和转录激活因子(JAK/STAT)信号通路;然而PCV3 Cap不影响STAT1和STAT2表达、磷酸化水平,以及STAT1和STAT2二聚体的形成;PCV3 Cap虽然能与KPNA1相互作用,但无法竞争阻断STAT1和KPNA1的结合,以及ISGF3复合体(激活的STAT1和STAT2与IRF9形成)入核;通过一系列pull-down和Co-IP试验发现,PCV3 Cap可能通过与STAT2羧基端的反式转录激活结构域(TAD)结合或与ISGF3的模拟物IRF9-S2C竞争结合ISRE从而抑制IFN-β诱导ISG的产生。以上研究结果表明,PCV3能通过Cap在多个途径抑制IFN-β介导的抗病毒免疫反应,具备调控宿主先天免疫反应和逃逸宿主抗DNA病毒反应的能力。

表1 与PCV3 Cap互作的宿主蛋白
2 PCV3-宿主蛋白的相互作用
作为单链DNA病毒,与PCV1和PCV2类似,PCV3 Cap蛋白可能参与其病毒生命周期的多个阶段。为深入了解PCV3在细胞内复制增殖的过程,Song等[29]在PK-15细胞转染表达PCV3 Cap蛋白和GFP融合蛋白,通过免疫沉淀结合LC-MS/MS质谱技术对宿主细胞内与PCV3 Cap互作的蛋白进行了分析,共鉴定出776个蛋白质,KEGG通路富集分析显示这些蛋白质主要涉及系统性红斑狼疮、坏死、血管平滑肌收缩、肾素分泌、病毒致癌、NOD样受体介导信号转导、促性腺激素释放激素信号转导、炎症介质调节瞬时受体电位通道等,推测系统性红斑狼疮、坏死、血管平滑肌收缩和肾素分泌相关通路可能与PCV3感染临床引起的猪皮炎肾病综合征有关。
PCV4于2019年在我国湖南省首次发现,临床症状与猪圆环病毒相关疾病相似,最近在中国和韩国的养殖场被检测到,被认为是威胁全球养猪业的潜在重要病原之一[30-34]。2022年,Zhou等[35]利用免疫共沉淀结合LC-MS质谱技术鉴定分析与PCV3或PCV4 Cap相互作用的宿主蛋白,并绘制了相互作用组图谱,结果显示,与PCV3 Cap潜在互作蛋白401个,其中有123个蛋白只与PCV3 Cap互作,能同时与PCV3和PCV4 Cap相互作用的蛋白共有278个;基因本体注释与通路富集分析表明,PCV3 Cap的交集互作蛋白主要参与细胞酰胺代谢、核酸结合、核糖核蛋白复合物结合和ATP依赖的RNA解旋酶活性等细胞信号通路。
目前,通过蛋白组学研究和免疫共沉淀技术验证了部分宿主蛋白能与PCV3 Cap结合互作,具体的互作蛋白见表1。
3 PCV3生命周期
3.1 参与PCV3生命周期的相关重要宿主因子
对PCV病毒生命周期的研究主要基于PCV1和PCV2,目前对PCV3生命周期和参与其中的细胞宿主蛋白知之甚少。在PCV2感染过程中,病毒粒子主要通过Cap与硫酸肝素、硫酸软骨素B以不对称方式的相互作用附着于宿主细胞表面[23,36],其入侵宿主细胞的机制主要有2种:(1)通过网格蛋白介导的胞吞作用或巨胞饮作用;(2)通过依赖Rho-GTP酶类调节肌动蛋白细胞骨架的方式[23]。然而PCV3 Cap蛋白与PCV2 Cap蛋白不同,其没有肝素结合基序XBBXBX(B是碱性氨基酸,X是中性或疏水性氨基酸),因此PCV3结合的受体蛋白有待进一步研究明确。2021年,Shi等[37]研究发现,PCV3能通过依赖发动蛋白-2和网格蛋白介导的内吞途径进入PK-15细胞,并且Rab5和Rab7在PCV3的运输和初级、次级内体定位中发挥了重要作用;内体酸化发生于病毒脱衣壳和释放基因组的过程,在PCV3感染前对细胞进行氯化铵处理,感染细胞数和Cap蛋白的表达显著下降。作为DNA病毒,PCV基因组的复制和转录,以及病毒粒子的装配均在细胞核内进行,PCV1和PCV2 Cap蛋白的核定位信号序列(NLS)通常由氮端碱性氨基酸组成,主要介导Cap向细胞核内运输,对病毒的复制和装配有重要意义。Mou等[38]通过对PCV3 Cap蛋白进行截短并分析其亚细胞定位情况从而鉴定出其NLS。核仁磷酸蛋白(NPM1)是一种多功能蛋白,其参与PCV2病毒生命周期。NPM1能与Cap蛋白结合,是病毒粒子入核的核转运受体[39]。2021年,Zhou等[40]研究发现,PCV3 Cap氮端的前34个氨基酸残基为其核仁定位信号(NoLS),NPM1通过PCV3 Cap的NoLS与之结合从而介导其入核,并且NMP1氮端结构域的第48位丝氨酸残基在其中发挥重要作用。根据以上研究,我们初步绘制了PCV3病毒生命周期的模式图(图1)。
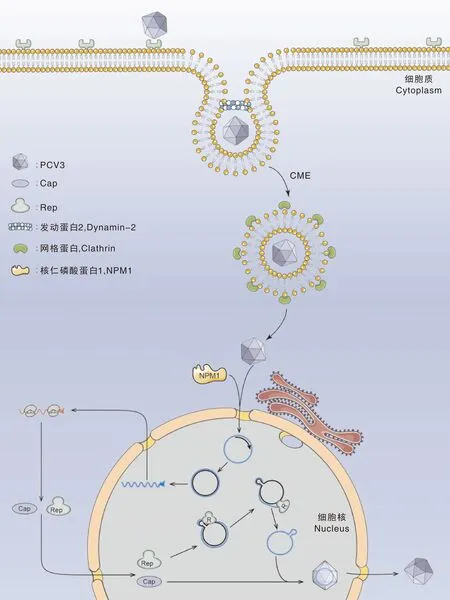
CME,网格蛋白介导的胞吞作用。
3.2 宿主因子对PCV3复制的影响
目前有关影响PCV3复制的宿主因子的报道仅有NPM1蛋白。Song等[29]发现,NMP1不仅能与PCV3 Cap相互作用,还能影响病毒复制;PCV3感染上调细胞内NPM1表达并且诱导其向核外转移至细胞质;过表达NPM1能够促进PCV3复制,细胞内病毒DNA拷贝数和Cap表达水平均上升;而下调NPM1表达则抑制PCV3的复制,表明NPM1正调控PCV3复制。根据上述结果,研究人员推测PCV3感染可能诱导NPM1结构和功能改变(包括mRNA转录、核糖体生物发生和蛋白质合成),而改变的NPM1会影响PCV3复制。NPM1调控PCV3复制的分子机制有待进一步探索。
4 展望
PCV3自发现以来就引起了全球学者的关注和兴趣,对PCV3的研究主要聚焦于病原微生物学、病原分子生物学和流行病学等方面,对其起源、遗传多样性和进化动态进行了一系列研究,但关于PCV3的生命周期与致病机制,目前知之甚少。病毒与宿主之间的相互作用决定了宿主细胞的命运和病毒的传播及存活。猪圆环病毒与宿主之间存在复杂的互作网络,其感染过程引起宿主细胞内环境改变,促使细胞演化出多种适应性应答机制以恢复稳态,而猪圆环病毒可以调控其中的一些信号通路以利于自身复制与增殖。PCV3感染可能引起宿主细胞自噬、凋亡和氧化应激,且对先天免疫与炎症反应有一定影响。一些宿主蛋白能与PCV3 Cap结合,参与病毒生命周期,或影响病毒复制。值得注意的是,临床症状涉及多系统炎症和引起的损伤与PCV3感染较为密切,可能与PCV3诱导的细胞凋亡和炎症相关信号通路有关。临床还发现,PCV3与其他猪病原体共感染的频率较高,这可能是由于PCV3引起免疫抑制导致其他病原体继发感染。后续研究可以对上述现象和推论进行探索,剖析相关分子机制。
