基于全基因组SNPs分析陆丰黄牛和雷琼牛的群体结构与遗传多样性特征
2023-08-14童雄罗威闵力张志飞马新燕罗成龙陈卫东徐斌李大刚
童雄,罗威,闵力,张志飞,马新燕,罗成龙,陈卫东,徐斌,李大刚
基于全基因组SNPs分析陆丰黄牛和雷琼牛的群体结构与遗传多样性特征
童雄,罗威,闵力,张志飞,马新燕,罗成龙,陈卫东,徐斌,李大刚
广东省农业科学院动物科学研究所/畜禽育种国家重点实验室/岭南现代农业科学与技术广东省实验室河源分中心/广东省畜禽育种与营养研究 重点实验室,广州 510640
【目的】研究陆丰黄牛和雷琼牛与世界不同地域家牛中的系统发育关系,解析不同家牛群体的遗传多样性,为地方家牛资源的鉴定与保护奠定理论基础。【方法】采集12头陆丰黄牛和17头雷琼牛的组织样品进行全基因组重测序,整合世界范围内分布的24个品种92个体的NCBI公共基因组数据,共计25个品种121个个体的信息开展群体遗传学研究。选用Bos taurus ARS-UCD1.2作为参考基因组,经基因组序列比对与质量控制获取高质量Reads,应用GATK软件检测基因组SNPs。进一步基于群体SNPs,利用系统进化树构建、PCA聚类和Admixture评估进行群体结构分析,通过核苷酸多样性()、杂合度()和连锁不平衡(LD)水平研究群体的遗传多样性。【结果】29头广东地方牛经基因组测序获取6 905 944 306个Clean Reads,每个样本平均基因组覆盖率为97.99%,平均测序深度分别为12.78×。整合NCBI公共基因组数据,经质控后共检测出14 664 391个群体SNPs。整合系统进化树、PCA、Admixture的结果发现普通牛与瘤牛分化,瘤牛群体存在中国瘤牛与印度瘤牛分化,普通牛群体中东北亚普通牛(韩牛、延边牛)和西藏牛与欧洲普通牛分化,温岭高峰牛和舟山牛从中国瘤牛群体中分化。陆丰黄牛和雷琼牛均属于纯正的中国瘤牛,但陆丰黄牛与皖南牛之间、雷琼牛与吉安牛之间呈现最近的亲缘关系,说明陆丰黄牛与地域临近的雷琼牛属于两个独立的品种。部分陆丰黄牛与雷琼牛均存在欧洲普通牛和东北亚普通牛的血统混杂,且混杂比例较高,说明这两个品种牛急需加强群体内的提纯复壮。相较于欧洲普通牛和韩牛,中国家牛群体的LD衰减速率更快,核苷酸多样性和杂合度更高,说明其遗传多样性更丰富。相较于其他中国家牛,陆丰黄牛和雷琼牛的LD水平更低,杂合度更高,且核苷酸多样性和杂合度的密度分布更为集中,说明两者受人工选择强度较低,且维持较高的群体遗传多样性。【结论】通过全基因组SNPs标记,系统解析了陆丰黄牛与雷琼牛的群体遗传结构和多样性特征,为这两个品种独立分类及其保护利用提供数据支撑。
陆丰黄牛;雷琼牛;全基因组SNPs;群体结构;遗传多样性
0 引言
【研究意义】家牛是人类驯化最早的家畜动物之一,距今约10 000年前由世界不同地域的原牛多次驯化而来[1-2]。中国家牛在长期的驯化和培育过程中受到人为选择和自然选择的作用,形成了表型丰富多样的地方品种[3],目前《国家畜禽遗传资源品种名录(2021年版)》收录的地方品种牛达55个,后续随着第三次全国畜禽遗传资源普查工作的推进,会有更多的地方品种牛被鉴定收录。陆丰黄牛和雷琼牛是广东省特有的地方品种,具有耐热、耐粗饲、抗病性强等优良特性[3-4],因此,这两个品种牛具有重要的开发利用价值和科学研究意义。近年来外来商业肉牛品种的引入导致这两个品种牛的种质资源流失,保种形势日趋严峻[4];同时,由于分子鉴定数据的缺乏,陆丰黄牛与雷琼牛在品种分类上是否归属于两个独立品种的问题,一直存在争议。【前人研究进展】《中国畜禽遗传资源志-牛志》在总结前人成果后,以生态类型、外貌特征、地理分布区域、分子生物学、生化遗传学等因素作为主要的分类依据,将中国黄牛划分为4种类型:华北型、中原型、南方型和培育品种[3],陆丰黄牛与雷琼牛从地域分布和表型特征上,属于典型的南方型中国黄牛[4]。前期基于线粒体mt-DNA序列[5]、常染色体基因组[6-7]和Y染色体基因组[7]分子标记开展的分类学研究,世界范围内家牛可分为2大类:瘤牛(Indicine, Bos taurues indicus)和普通牛(Taurine, Bos taurus taurus),进一步细分可分为5类型:欧亚普通牛(Eurasian taurine)、东亚普通牛(East Asian taurine)、欧洲普通牛(European taurine)、中国瘤牛(Chinese indicine)和印度瘤牛(Indian indicine)[7]。中国家牛主要来源于3个祖代类群:欧亚普通牛、东亚普通牛和中国瘤牛,中国西北部家牛属于欧亚普通牛,中国南方家牛属于中国瘤牛,中国中北部和西南部家牛均属于普通牛和瘤牛的杂交类群,西藏地区家牛属于东亚普通牛[7]。前期常染色体基因组SNPs评估遗传多样性研究中均反映出中国家牛的遗传多样性高于印度瘤牛,而两者高于欧洲普通牛;中国家牛中南方瘤牛的遗传多样性要高于北方的普通牛[7-8]。与此相反,mt-DNA单倍型多态性研究发现中国北方普通牛的遗传多样性较南方瘤牛更为丰富[5]。前期血液同工酶多态性研究报道了雷琼牛与文山牛和温岭高峰牛等南方瘤牛亲缘关系相近,且雷琼牛的群体遗传多样性丰富,高于外来商用品种牛[9]。基于mt-DNA序列的母系遗传学研究则报道雷琼牛mt-DNA的遗传多样性较低[10],在分类上属于瘤牛,但其特有的单倍型与印度瘤牛存在显著差异[11-12]。近年来,通过全基因组SNPs进行群体遗传学研究中,陆续报道雷琼牛具有纯正的中国瘤牛血统,且中国家牛中混杂的瘤牛血统均来自雷琼牛为代表的中国瘤牛而不是印度瘤牛[7-8]。有关陆丰黄牛的群体遗传学研究较少,目前LIU等在研究8个中国南方家牛品种的群体基因组特征时,发现陆丰黄牛与其他7个南方牛品种的遗传距离较远,且存在一定比例的特异性祖代血统[13],但该项研究中仅整合南方少数的几个牛品种,缺乏其他地域家牛的信息,因此,无法判定采集的陆丰黄牛样本是否存在遗传混杂,进而引发群体结构解析偏差。此外,LIU等先后利用该样本数据,比较中国南方牛群体遗传多样性时,发现陆丰黄牛和海南牛(海南雷琼牛群体)近交系数较其他南方牛(文山牛、广东雷琼牛群体等)高,且其遗传多样性更低[13-14]。【本研究切入点】近年来随着高通量测序技术的发展,世界各地域分布的家牛陆续开展基因组学研究[6-8, 15-18],目前通过整合世界各地域代表性牛品种的基因组信息,将是系统解析陆丰黄牛和雷琼牛群体遗传结构和遗传多样性的契机。【拟解决的关键问题】本研究采集陆丰黄牛和雷琼牛的群体样本进行全基因组测序,整合其他地域代表性牛品种的基因组数据,解析陆丰黄牛与雷琼牛之间、两者与其他地域牛品种之间的亲缘关系,指导品种的分类鉴定。同时,不同家牛群体的遗传多样性分析,为后续地方品种保种方案制定提供信息支持。
1 材料与方法
1.1 样品采集与数据库信息收集
研究于2022年6—7月,分别在广东省陆丰市潭西镇陆丰黄牛保种场和广东省湛江市麻章区雷琼牛保种场采集12头陆丰黄牛(图1)的耳组织样和17头雷琼牛(图2)的外周血样,进行基因组DNA提取、质量检测、文库构建与测序。为解析广东2个地方品种牛与世界其他地域家牛品种之间的系统发育关系,本研究从NCBI数据库下载24个品种92个个体的全基因组重测序数据,所有数据库个体的下载信息见附表1。本研究25个品种121个个体的样本信息详见表1。

图2 雷琼牛(公)
1.2 试验方法
1.2.1 基因组DNA提取与质量检测 采用传统的苯酚-氯仿法进行组织样基因组DNA的抽提。根据Illumina NovaSeq6000系统(Illumina, Inc., San Diego, CA, USA)对基因组DNA样品的文库构建质量要求:OD260/280介于1.8—2.0之间,DNA总量≥10μg。
1.2.2 基因组文库构建与测序 29份组织样品检测合格后,每个样品取用0.5μg DNA模板,根据Annoroad® Universal DNA Fragmentase kit V2.0(AN200101-L)及Annoroad® Universal DNA Library Prep Kit V2.0(AN200101-L)的方法及流程进行文库制备。使用NovaSeq 6000 S4 Reagent kit V1.5试剂在NovaSeq 6000 S4平台进行成簇和测序,运行双端测序程序(PE),得到150 bp的双端测序Reads。
1.3 数据处理
1.3.1 基因组序列比对与质量控制 将原始数据(Raw Data)中带接头和低质量的序列进行过滤,从而得到高质量序列(Clean Reads)。过滤步骤如下:(1)去除接头污染的Reads(Reads中接头污染的碱基数大于5 bp);(2)去除低质量的Reads(超过50%的碱基质量值低于19的Reads);(3)去除含N比例大于5%的Reads。
选用Bos taurus ARS-UCD1.2[19]作为参考基因组,通过基因组比对软件BWA v.0.7.17[20]将CleanReads比对到参考基因组上。利用Samtools v.1.9软件[21]对比对后的序列进行排序,且通过-q 4参数去除低质量Reads,应用Picard-tools v.2.20.2软件(http:// broadinstitute.github.io/picard/)去除PCR重复。
1.3.2 基因组SNPs的检测 应用GATK v.3.8软件[22]中检测群体位点变异的方法检测SNP。通过质量值、深度、重复性等条件进行过滤筛选,过滤参数设置为:QD<2.0, ReadPosRankSum<-8.0, FS>60.0, QUAL< 30.0, DP<4.0, MQ<40.0, MappingQualityRankSum<-12.5。利用VCFtools v0.1.16软件[23]提取常染色体SNPs后,以参数“--min-alleles 2 --max-alleles 2 --maf 0.05 --max- missing 0.9 --minGQ 15.0”进行质控,获得群体高质量常染色体SNPs。
1.3.3 群体遗传结构分析 系统进化树分析:利用Plink v1.9软件[24]计算群体IBS遗传距离,通过MEGA v8.0软件[25]构建Neighbor-joining tree。PCA分析:应用Plink v1.9软件[24]进行LD-pruning,剔除高连锁群体SNPs后,利用Eigenstrat v6.1.4软件[26]进行PCA分析。Admixture分析:基于LD-pruning后的群体SNPs,使用Admixture v1.3.0软件[27]的 unsupervised mode进行Admixture分析,计算K=2-5的群体遗传组分。
1.3.4 群体内遗传多样性分析 杂合度分析:自制perl脚本运行分析,bin size设为40 000,step window size设为20 000。核苷酸多样性分析:利用VCFtools v0.1.16软件[23]进行计算,参数设置:“--window-pi 40000,--window-pi-step 20000”。连锁不平衡LD分析:使用PopLDdecay V3.40软件[28],对各群体的LD decay进行统计与作图。
2 结果
2.1 基因组数据比对质量与数据合并
本研究采集29头广东地方牛样品进行基因组测序,获得6 905 944 306个Clean Reads,每个样本平均基因组覆盖率为97.99%,平均测序深度分别为12.78×。将本研究采集的29个样本的基因组测序数据与NCBI数据库下载的24个品种92个个体的基因组测序数据进行合并分析(表1),检测出14 664 391个群体SNPs。基于质控条件设置SNP缺失率≤0.15,故LF08个体在后续研究中被去除。
2.2 不同地域分布家牛的群体遗传结构
2.2.1 系统进化树 基于状态一致性(Identity By State, IBS)距离构建全群的邻近进化树,推断不同品种牛之间的系统发育关系(图3)。本研究采集的雷琼牛与NCBI数据库下载的3头雷琼牛聚为一簇,这说明本研究采集样本具有品种代表性,且邻近进化树构建方法科学合理。进化树上清晰反映:所有家牛出现普通牛(安格斯牛、海福特牛、利木赞牛、西门塔尔牛、西藏牛、韩牛、延边牛、鲁西牛、渤海黑牛、南阳牛)和瘤牛(锦江牛、皖南牛、广丰牛、吉安牛、雷琼牛、陆丰黄牛、文山牛、舟山牛、温岭高峰牛、哈里亚娜牛、吉尔牛、沙希华牛、内洛尔牛、塔帕卡牛、婆罗门牛)两个大类群;在瘤牛群体中,中国瘤牛(锦江牛、皖南牛、广丰牛、吉安牛、雷琼牛、陆丰黄牛、文山牛、舟山牛、温岭高峰牛)与印度瘤牛(哈里亚娜牛、吉尔牛、沙希华牛、内洛尔牛、塔帕卡牛、婆罗门牛)形成了两个独立的类群。此外,在中国瘤牛群体中,浙江省的舟山牛和温岭高峰牛形成一个相对独立的类群。陆丰黄牛和雷琼牛均属于中国瘤牛类群,雷琼牛群体内仅1个个体(Leiqiong12)偏离,其他个体均能聚为一簇;而陆丰黄牛群体内出现2个亚群(亚群1:lufeng01、lufeng04、lufeng07、lufeng10和lufeng12;亚群2:lufeng02、lufeng05、lufeng06和lufeng09),且亚群外的2个个体(Lufeng03和Lufeng11)遗传距离与中国北方牛(南阳牛和鲁西牛)相近。
2.2.2 主成分分析(PCA) 对120个个体进行主成分分析,计算特征值的TW检验统计量,并对TW进行显著性判定。前2个显著性特征向量PC1和PC2分别解释16.19%和3.44%的变异。通过PC1和PC2建立的二维PCA图(图4)中:PC1分布可解释普通牛和瘤牛之间存在显著的遗传分化;PC2分布解释了中国瘤牛与印度瘤牛之间的遗传差异,且陆丰黄牛与雷琼牛均归属于中国瘤牛类群,这与系统进化树的结果相一致。

表1 本研究中25个品种121个个体的样本信息

Angus:安格斯牛;Hereford:海福特牛;Limousin:利木赞牛;Simmental:西门塔尔牛;Tibetan:西藏牛;Hanwoo:韩牛;Yanbian:延边牛;Luxi:鲁西牛;Bohai:渤海黑牛;Nanyang:南阳牛;Jinjiang:锦江牛;Wannan:皖南牛;Guangfeng:广丰牛;Jian:吉安牛;Leiqiong:雷琼牛;Lufeng:陆丰黄牛;Wenshan:文山牛;Zhoushan:舟山牛;Wenling:温岭高峰牛;Hariana:哈里亚娜牛;Gir:吉尔牛;Sahiwal:沙希华牛;Nelore:内洛尔牛;Tharparkar:塔帕卡牛;Brahman:婆罗门牛。品种代号后的数字代表品种内个体的编号。下同
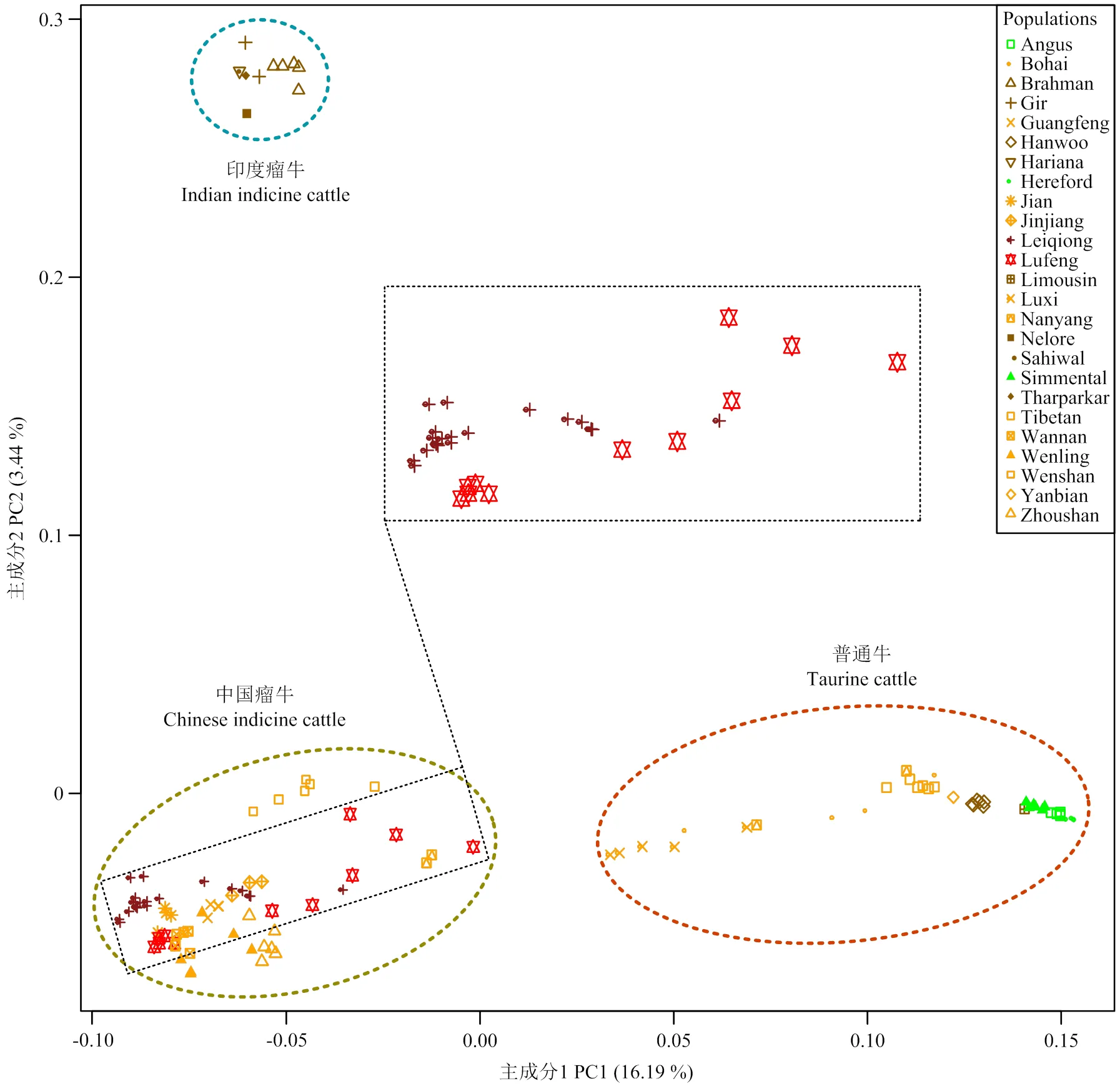
图中虚线方框中仅显示陆丰黄牛和雷琼牛的PCA。The dotted box in the figure shows only the PCA of Lufeng cattle and Leqiong cattle.
2.2.3 Admixture祖代分析 Admixture评估家牛群体的祖代数量和遗传混杂比例,随着祖代群体数K的逐步递增(K=2-5),所研究样本群体呈现不同的群体结构特征(图5)。当K=2-3时,普通牛和瘤牛分离,瘤牛群体分化为:中国瘤牛和印度瘤牛,这与PCA和系统进化树的结果一致。K=4时,西藏牛、韩牛、延边牛与欧洲普通牛出现明显的分化。K=5时,温岭高峰牛和舟山牛从中国瘤牛中分离出来,这与系统进化树的结果一致。5个陆丰黄牛和12个雷琼牛具有单一的祖代成分,而剩余的6个陆丰黄牛和8个雷琼牛均存在多个祖代成分,且主要受到欧洲普通牛和东亚普通牛的混杂影响。5个单一祖代成分的陆丰黄牛与系统进化树中亚群1个体完全一致。
2.3 不同地域分布家牛的核苷酸多样性和杂合度分析
对18个家牛品种(海福特牛、安格斯牛、西门塔尔牛、韩牛、西藏牛、渤海黑牛、鲁西牛、南阳牛、舟山牛、婆罗门牛、温岭高峰牛、文山牛、雷琼牛、陆丰黄牛、广丰牛、吉安牛、皖南牛、锦江牛)进行种群核苷酸多样性()和杂合度()评估(图6)。除西藏牛外,中国家牛的核苷酸多样性和杂合度的中位数与婆罗门牛相近,但均高于欧洲普通牛(海福特牛、安格斯牛、西门塔尔牛)和韩牛。欧洲普通牛和韩牛核苷酸多样性的四分位距较中国家牛小,说明核苷酸多样性较中国家牛更为集中。欧洲普通牛和韩牛核苷酸多样性和杂合度分布不均匀,密度分布集中中位数与下四分位数之间,说明欧洲普通牛和韩牛中低的多样性和杂合度值较集中分布。陆丰黄牛和雷琼牛和的四分位距较其他中国家牛小,说明和较其他中国家牛更为集中。此外,这两个品种牛的中位数高于其他中国家牛,且密度分布的峰值更高宽度更窄,说明其较其他中国家牛更高,且变异程度小。

品种代号后的数字代表品种内个体的编号 The number after breed code represents the number of the individual within the breed.
2.4 不同地域分布家牛的连锁不平衡分析
为评估18个家牛群体的连锁不平衡水平,应用全基因组SNPs绘制不同群体的2衰减图(图7)。相比欧洲普通牛(海福特牛、安格斯牛、西门塔尔牛)和韩牛,除西藏牛外,中国家牛(渤海黑牛、鲁西牛、南阳牛、舟山牛、温岭高峰牛、文山牛、雷琼牛、陆丰黄牛、广丰牛、吉安牛、皖南牛、锦江牛)和婆罗门牛群体的LD衰减速率更快。陆丰黄牛和雷琼牛群体的LD水平(2值)较其他中国家牛群体低。
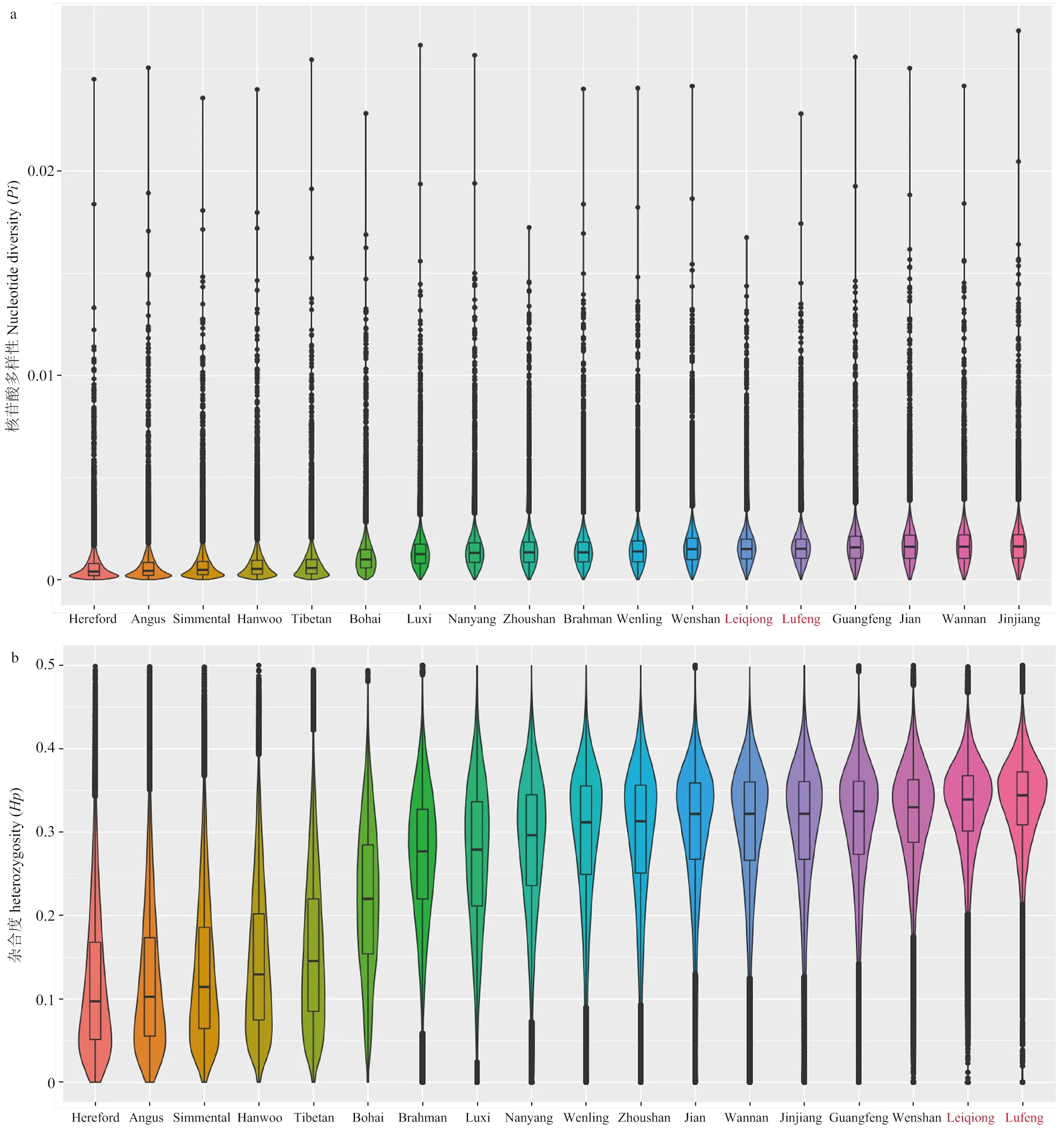
图6 不同家牛群体的核苷酸多样性(a,Pi)和杂合度(b,Hp)
3 讨论
3.1 不同地域分布家牛的群体遗传结构
现代家牛均由共同的祖先-原牛驯化而来,前期mt-DNA遗传方面的研究发现普通牛和瘤牛来源于两个独立分化的原牛群体,且两者在母系遗传分化的时间约为20万年前[32]。本研究中系统进化树、PCA和Admixture分析结果一致显示:普通牛类群和瘤牛类群存在明显的遗传分化,这与之前基因芯片[6, 16, 33]和基因组重测序研究[7, 17]中得到的结果一致,从基因组水平上反映出:世界范围内不同地域分布的家牛主要由普通原牛和瘤原牛分别驯化而来。前期考古学的研究表明,中国南方瘤牛是距今约3 500—2 500年前由印度次大陆的瘤牛迁移扩散而来[34]。CHEN等[7]和WANG等[16]从常染色体和Y染色体水平上均发现瘤牛群体分化为中国瘤牛和印度瘤牛,且推断出两者分歧时间约为3.66万—4.96万年,早于瘤牛驯化时间。本研究增补4个中国南方瘤牛品种(陆丰黄牛、雷琼牛、舟山牛、温岭高峰牛)的基因组变异信息,同样证实了两个瘤牛类群之间的分化,综合前期考古和分子遗传学的研究成果,造成遗传分化的原因可能是两个类群具有独立的祖先。本研究整合了CHEN等[7]研究中部分样本,因此同样发现西藏牛与东北亚的普通牛(韩牛、延边牛)遗传距离相近,而与欧洲普通牛存在明显的遗传分化。品种形成历史上,舟山牛是由上海川沙、南汇等地的牛只引入后形成的品种[3, 35],而温岭高峰牛是在当地现有牛群的基础上选育而来[3]。JIANG等在研究舟山牛与其他品种牛之间的亲缘关系时,发现舟山牛与温岭高峰牛的亲缘关系相近,且两个品种牛与相邻地域的中国瘤牛(皖南牛、吉安牛、雷琼牛)形成明显的遗传分化[15]。本研究整合欧洲普通牛、印度瘤牛及更多的中国瘤牛品种(n=9),同样发现这两个品种牛遗传距离相近,且从中国瘤牛类群中分离,但部分温岭高峰牛个体存在祖代血统混杂,这说明舟山牛可能在相对封闭地域环境下形成了特有的群体,而温岭高峰牛在近期可能受到舟山牛和其他中国瘤牛基因交流的共同影响。
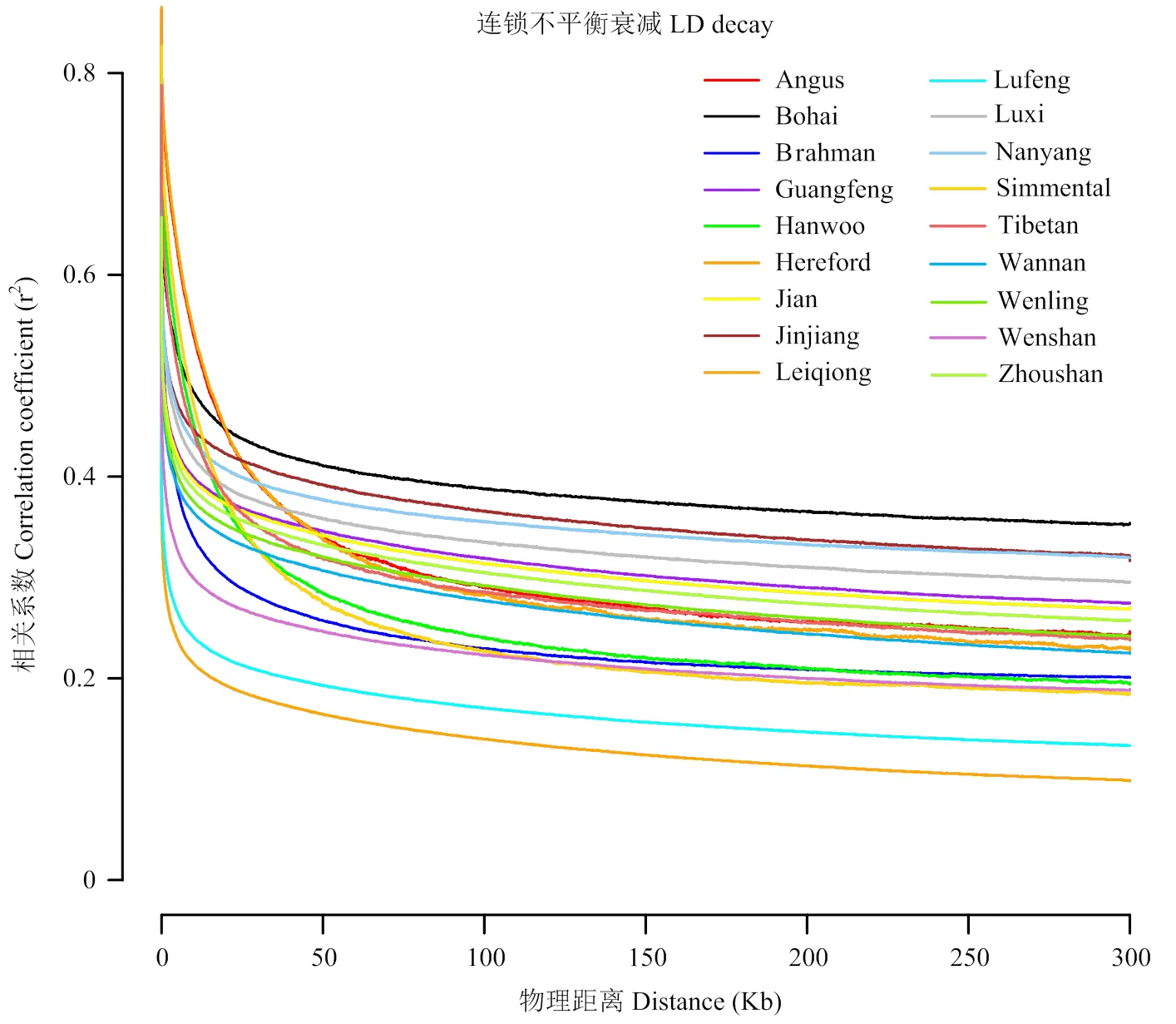
图7 不同家牛群体的连锁不平衡LD衰减
3.2 陆丰黄牛和雷琼牛与其他地域家牛品种间的亲缘关系
通过采集广东陆丰黄牛和雷琼牛的群体样本,整合NCBI数据库公布的24个品种重测序数据,发现广东2个地方品种牛均属于中国瘤牛类群。本研究系统进化树和Admixture分析发现5个血统纯正的陆丰黄牛个体与地理分布相对较远的皖南牛呈现最近的亲缘关系,这可能是自古徽商与粤商密集商贸往来而引入的基因交流产生的结果,皖南牛核心产区所在的歙县等地正是徽商主要的发源地,而陆丰黄牛主要分布的粤东地区是粤商商贸的代表性区域[4, 36]。进一步从Admixture遗传混杂比例结果来看,未受到遗传混杂影响的陆丰黄牛与雷琼牛具有单一纯正的祖代,皖南牛则主要来源于广东来源的瘤牛祖代血统,同时,受到其他中国瘤牛祖代的遗传混杂影响,因此,可以推测陆丰黄牛在商贸往来介导之下,单向流入皖南地区进而影响皖南牛的品种形成。CHEN等在研究不同地域家牛祖代特征时发现皖南牛与雷琼牛具有相同的单一祖代[7],而本研究在整合舟山牛、温岭高峰牛和陆丰黄牛后,发现皖南牛存在多种中国瘤牛血统混杂的情况,进而解析皖南牛的品种形成及其与陆丰黄牛的亲缘关系。本研究系统进化树反映出雷琼牛和吉安牛的亲缘关系最近,这与CHEN等[7,15-16]的研究结果一致,结合Admixture遗传混杂结果可以发现吉安牛主要血统来自雷琼牛为代表的祖代,且混杂了少量舟山牛和温岭高峰牛来源的祖代及欧亚普通牛祖代,因此,推断自古广东与江西的行政管理和贸易流通[37]介导了雷琼牛与吉安牛相互影响,而吉安牛广袤的中心产区(江西省吉安市、赣州市、抚州市、湖南省茶陵、衡山)[3]易受邻近地区和外来牛种的基因渗入影响。前期血液同工酶多态性研究发现,相比于其他地域家牛,雷琼牛与中国南方瘤牛(文山牛和温岭高峰牛)亲缘关系更近[9];而基于mt-DNA序列开展雷琼牛母系起源研究中报道了雷琼牛具有特有的单倍型,与印度瘤牛之间存在遗传差异[11-12]。CHEN等[7-8]在家牛群体基因组研究中报道中国南方瘤牛可能是独立驯化而来,雷琼牛具有纯正的中国瘤牛血统,且中国家牛中瘤牛血统来自雷琼牛为代表的中国瘤牛而不是印度瘤牛,这与本研究系统进化树、PCA和Admixture分析结果一致。本研究中部分陆丰黄牛与雷琼牛均存在欧洲普通牛和东亚普通牛的血统混杂,且混杂比例较高,这说明广东地方品种牛在近期可能受到外来品种牛的基因渗入影响,急需加强品种内的提纯复壮。
3.3 不同地域家牛的遗传多样性比较
通过核苷酸多样性和杂合度的中位数及数据分布结果均反映出中国家牛的遗传多样性与印度瘤牛相近,而均高于欧洲普通牛(海福特牛、安格斯牛、西门塔尔牛)和韩牛,这可能与欧洲普通牛和韩牛作为商业品种牛经过高度选育有关。同时,本研究发现中国家牛和婆罗门牛群体的LD衰减速率快于欧洲普通牛和韩牛,这与XIA等[38-39]报道的LD衰减趋势一致,同时也验证了中国家牛和婆罗门牛具有更高的遗传多样性。本研究Admixture的结果反映出中国北方家牛和部分南方家牛存在广泛的多祖代遗传混杂,这可能是维持中国家牛群体较高的遗传多样性的重要来源。前期中国黄牛基因组研究中也陆续报道中国家牛中广泛的基因交流,进而影响了秦川牛[8]、滇中牛[7]、涠洲黄牛[16]、大别山牛[40]等群体的遗传多样性。北方家牛中欧洲普通牛的血统比例较中国南方家牛更高,这与CHEN等[7,16]报道的一致。广东2个地方品种牛较其他中国家牛具有更为集中的核苷酸多样性和杂合度,笔者结合Admixture的结果推断这可能是广东2个品种受人工选择作用的影响较低。此外,广东2个地方品种牛具有较高的杂合度,高于文山牛等其他中国家牛品种,这可能是本研究选取的参考基因组与其遗传距离较远所产生或是群体高遗传多样性的体现。从中国家牛群体的LD水平来看,广东2个地方品种牛的LD水平明显低于其他中国家牛,这进一步说明这2个品种牛具有高的遗传多样性,且受人工选择的强度较低。与本研究中杂合度和LD的趋势正好相反,LIU等先后利用芯片技术研究中国南方家牛的群体遗传多样性时,发现相比文山牛、雷琼牛(广东雷琼牛群体)、昭通牛等5个南方牛品种,陆丰黄牛与海南牛(海南雷琼牛群体)具有更低的杂合度(和)和更高的近交系数()[13-14],这可能与不同研究中采集样本的群体结构与多样性有关。杨关福等在研究雷琼牛血液蛋白的遗传多样性时,发现雷琼牛的多态座位百分比和平均杂合度较高,且均高于荷斯坦牛[41],这与本研究中雷琼牛的遗传多样性高于欧洲普通牛的趋势是一致的。与此相反,前期mt-DNA的遗传多样性研究中,雷琼牛D-loop区的核苷酸多样性仅为0.15%,低于秦川牛、蒙古牛、中国荷斯坦牛等品种[10]。
4 结论
本研究采集29头广东地方牛(12头陆丰黄牛和17头雷琼牛),整合24个品种92个体的NCBI公共基因组数据,开展陆丰黄牛和雷琼牛的遗传多样性与群体结构特征研究。陆丰黄牛和雷琼牛均属于纯正的中国瘤牛,陆丰黄牛与皖南牛之间、雷琼牛与吉安牛之间呈现最近的亲缘关系。中国家牛群体相较于欧洲普通牛和韩牛,具有更快的LD衰减速率和更高的核苷酸多样性和杂合度相比于其他中国家牛,陆丰黄牛和雷琼牛LD水平更低,杂合度更高,核苷酸多样性和杂合度的密度分布更为集中。部分陆丰黄牛与雷琼牛均存在较高比例的欧洲普通牛和东北亚普通牛的血统混杂,在后续的品种保护中急需进行群体内的提纯复壮。
[1] LOFTUS R T, MACHUGH D E, BRADLEY D G, SHARP P M, CUNNINGHAM P. Evidence for two independent domestications of cattle. Proceedings of the National Academy of Sciences of the United States of America, 1994, 91(7): 2757-2761.
[2] HESSE B. The first steps of animal domestication. Journal of Ethnobiology, 200626(1): 171-174.
[3] 国家畜禽遗传资源委员会组. 中国畜禽遗传资源志-牛志. 北京: 中国农业出版社, 2011.
China National Commission of Animal Genetic Resources. Animal genetic resources in China. Beijing: China Agriculture Press, 2011. (in Chinese)
[4] 广东省农业厅, 广东省畜牧兽医局, 广东省畜禽遗传资源委员会. 广东省地方畜禽遗传资源志. 广州: 广东科技出版社, 2018.
Department of Agriculture of Guangdong Province. Records of local livestock and poultry genetic resources in Guangdong Province. Guangzhou: Guangdong Science & Technology Press, 2018. (in Chinese)
[5] XIA X, QU K, ZHANG G, JIA Y, MA Z, ZHAO X, HUANG Y, CHEN H, HUANG B, LEI C. Comprehensive analysis of the mitochondrial DNA diversity in Chinese cattle. Animal Genetics, 2019, 50(1): 70-73.
[6] GAO Y H, GAUTIER M, DING X D, ZHANG H, WANG Y C, WANG X, FARUQUE M O, LI J Y, YE S H, GOU X, HAN J L, LENSTRA J A, ZHANG Y. Species composition and environmental adaptation of indigenous Chinese cattle. Scientific Reports, 2017, 7(1): 1-14.
[7] CHEN N B, CAI Y D, CHEN Q M, LI R, WANG K, HUANG Y Z, HU S M, HUANG S S, ZHANG H C, ZHENG Z Q, et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nature Communications, 2018, 9(1): 1-13.
[8] MEI C G, WANG H C, LIAO Q J, WANG L Z, CHENG G, WANG H B, ZHAO C P, ZHAO S C, SONG J Z, GUANG X M, et al. Genetic architecture and selection of Chinese cattle revealed by whole genome resequencing. Molecular Biology and Evolution, 2018, 35(3): 688-699.
[9] 俞英, 文际坤, 朱芳贤, 赵开典, 宿兵, 王文, 张亚平, 刘爱华, 季维智, 施立明. 云南文山黄牛和迪庆黄牛遗传多样性的蛋白电泳研究. 动物学研究, 1997, 18(3): 333-339.
YU Y, WEN J K, ZHU F X, ZHAO K D, SU B, WANG W, ZHANG Y P, LIU A H, JI W Z, SHI L M. Genetic diversity of Wenshan and Diqing yellow cattle in Yunan province assayed by protein electrophoresis. Zoological Research, 1997, 18 (3): 333-339. (in Chinese)
[10] 王朝锋, 雷初朝, 陈宏, 蔡欣, 党瑞华, 欧江涛. 雷琼牛mtDNA D—loop遗传多态性研究. 黄牛杂志, 2005(5): 14-15.
WANG C F, LEI C C, CHEN H, CAI X, DANG R H, OU J T. Study on mtDNA D-loop genetic diversity in Leiqiong cattle. Journal Yellow Cattle Science, 2005(5): 14-15. (in Chinese)
[11] YU Y, NIE L, HE Z Q, WEN J K, JIAN C S, ZHANG Y P. Mitochondrial DNA variation in cattle of South China: origin and introgression. Animal Genetics, 1999, 30(4): 245-250.
[12] 耿荣庆, 常洪, 冀德君, 王兰萍, 常国斌, 李世平, 马国龙, 陈宏宇, 常春芳, 李永红. 雷琼牛母系起源的遗传学证据. 畜牧兽医学报, 2008, 39(7)849-852.
GENG R Q, CHANG H, JI D J, WANG L P, CHANG G B, LI S P, MA G L, CHEN H Y, CHANG C F, LI Y H. Genetic evidence of maternal origin of Leiqiong cattle. Acta Veterinaria et Zootechnica Sinica, 2008, 39(7)849-852. (in Chinese)
[13] LIU Y Q, XU L Y, YANG L, ZHAO G Y, LI J Y, LIU D W, LI Y K. Discovery of genomic characteristics and selection signatures in southern Chinese local cattle. Frontiers in Genetics, 2020, 11: 533052.
[14] LIU Y Q, ZHAO G Y, LIN X J, ZHANG J H, HOU G Y, ZHANG L P, LIU D W, LI Y K, LI J Y, XU L Y. Genomic inbreeding and runs of homozygosity analysis of indigenous cattle populations in Southern China. PLoS ONE, 2022, 17(8): e0271718.
[15] JIANG L H, KON T, CHEN C Y, ICHIKAWA R, ZHENG Q Y, PEI L Y, TAKEMURA I, NSOBI L H, TABATA H, PAN H, OMORI Y, OGURA A. Whole-genome sequencing of endangered Zhoushan cattle suggests its origin and the association of MC1R with black coat colour. Scientific Reports, 2021, 11(1): 1-11.
[16] WANG X G, JU Z H, JIANG Q, ZHONG J F, LIU C K, WANG J P, HOFF J L, SCHNABEL R D, ZHAO H, GAO Y P, et al. Introgression, admixture, and selection facilitate genetic adaptation to high-altitude environments in cattle. Genomics, 2021, 113(3): 1491-1503.
[17] KIM K, KWON T, DESSIE T, YOO D, MWAI O A, JANG J, SUNG S, LEE S, SALIM B, JUNG J, et al. The mosaic genome of indigenous African cattle as a unique genetic resource for African pastoralism. Nature Genetics, 2020, 52(10): 1099-1110.
[18] 宋娜娜, 钟金城, 柴志欣, 汪琦, 何世明, 吴锦波, 蹇尚林, 冉强, 蒙欣. 三江黄牛全基因组数据分析. 中国农业科学, 2017, 50(1): 183-194.
SONG N N, ZHONG J C, CHAI Z X, WANG Q, HE S M, WU J B, JIAN S L, RAN Q, MENG X. The whole genome data analysis of Sanjiang cattle. Scientia Agricultura Sinica, 2017, 50(1): 183-194. (in Chinese)
[19] ROSEN B D, BICKHART D M, SCHNABEL R D, KOREN S, ELSIK C G, TSENG E, ROWAN T N, LOW W Y, ZIMIN A, COULDREY C, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience, 2020, 9(3): giaa021.
[20] LI H, DURBIN R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009, 25(14): 1754-1760.
[21] LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, MARTH G, ABECASIS G, DURBIN R, SUBGROUP 1 G P D P. The sequence alignment/map format and SAMtools. Bioinformatics, 2009, 25(16): 2078-2079.
[22] MCKENNA A, HANNA M, BANKS E, SIVACHENKO A, CIBULSKIS K, KERNYTSKY A, GARIMELLA K, ALTSHULER D, GABRIEL S, DALY M, DEPRISTO M A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 2010, 20(9): 1297-1303.
[23] DANECEK P, AUTON A, ABECASIS G, ALBERS C A, BANKS E, DEPRISTO M A, HANDSAKER R E, LUNTER G, MARTH G T, SHERRY S T, MCVEAN G, DURBIN R, GROUP 1 G P A. The variant call format and VCFtools. Bioinformatics, 2011, 27(15): 2156-2158.
[24] PURCELL S, NEALE B, TODD-BROWN K, THOMAS L, FERREIRA M A R, BENDER D, MALLER J, SKLAR P, DE BAKKER P I W, DALY M J, SHAM P C. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics, 2007, 81(3): 559-575.
[25] KUMAR S, STECHER G, LI M, KNYAZ C, TAMURA K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 2018, 35(6): 1547-1549.
[26] PATTERSON N, PRICE A L, REICH D. Population structure and eigenanalysis. PLoS Genetics, 2006, 2(12): e190.
[27] ALEXANDER D H, NOVEMBRE J, LANGE K. Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 2009, 19(9): 1655-1664.
[28] ZHANG C, DONG S S, XU J Y, HE W M, YANG T L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics, 2019, 35(10): 1786-1788.
[29] STOTHARD P, LIAO X P, ARANTES A S, DE PAUW M, COROS C, PLASTOW G S, SARGOLZAEI M, CROWLEY J J, BASARAB J A, SCHENKEL F, MOORE S, MILLER S P. A large and diverse collection of bovine genome sequences from the Canadian Cattle Genome Project. GigaScience, 2015, 4(1): s13742-15.
[30] SHIN D H, LEE H J, CHO S, KIM H J, HWANG J Y, LEE C K, JEONG J, YOON D, KIM H. Deleted copy number variation of Hanwoo and Holstein using next generation sequencing at the population level. BMC Genomics, 2014, 15: 240.
[31] BICKHART D M, XU L Y, HUTCHISON J L, COLE J B, NULL D J, SCHROEDER S G, SONG J Z, GARCIA J F, SONSTEGARD T S, VAN TASSELL C P, et al. Diversity and population-genetic properties of copy number variations and multicopy genes in cattle. DNA Research, 2016, 23(3): 253-262.
[32] ACHILLI A, BONFIGLIO S, OLIVIERI A, MALUSÀ A, PALA M, KASHANI B H, PEREGO U A, AJMONE-MARSAN P, LIOTTA L, SEMINO O, et al. The multifaceted origin of taurine cattle reflected by the mitochondrial genome. PLoS One, 2009, 4(6): e5753.
[33] DECKER J E, MCKAY S D, ROLF M M, KIM J, MOLINA ALCALÁ A, SONSTEGARD T S, HANOTTE O, GÖTHERSTRÖM A, SEABURY C M, PRAHARANI L, et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genetics, 2014, 10(3): e1004254.
[34] CAI D W, SUN Y, TANG Z W, HU S M, LI W Y, ZHAO X B, XIANG H, ZHOU H. The origins of Chinese domestic cattle as revealed by ancient DNA analysis. Journal of Archaeological Science, 2014, 41: 423-434.
[35] CHEN Y C. Four interesting endangered breeds of animals in China. Animal Genetic Resources Information, 1995, 16: 29-35.
[36] 魏霞, 刘正刚. 明清安徽与广东的贸易往来. 安徽史学, 2001(4): 18-21.
WEI X, LIU Z G. On the trade of Anhui Province with Guangdong Province during the Ming and Qing dynasties. Historiography Anhui, 2001(4): 18-21. (in Chinese)
[37] 王海. 明清粤赣通道与两省毗邻山地互动发展研究[D]. 广州: 暨南大学, 2008.
WANG H. Study on the interactive development of Guangdong- jiangxi passage and the adjacent mountainous areas of the two provinces in Ming and Qing dynasties[D]. Guangzhou: Jinan University, 2008. (in Chinese)
[38] XIA X T, ZHANG S J, ZHANG H J, ZHANG Z J, CHEN N B, LI Z G, SUN H X, LIU X, LYU S J, WANG X W, LI Z M, YANG P, XU J W, DING X T, SHI Q T, WANG E Y, RU B R, XU Z J, LEI C Z, CHEN H, HUANG Y Z. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genomics, 2021, 22(1): 43.
[39] ZHANG S J, YAO Z, LI X M, ZHANG Z J, LIU X, YANG P, CHEN N B, XIA X T, LYU S J, SHI Q T, et al. Assessing genomic diversity and signatures of selection in Pinan cattle using whole-genome sequencing data. BMC Genomics, 2022, 23(1): 460.
[40] GUAN X W, ZHAO S P, XIANG W X, JIN H, CHEN N B, LEI C Z, JIA Y T, XU L. Genetic diversity and selective signature in Dabieshan cattle revealed by whole-genome resequencing. Biology, 2022, 11(9): 1327.
[41] 杨关福, 张细权, 李加琪, 聂龙, 王文. 徐闻黄牛和海南黄牛血液蛋白的遗传多样性. 华南农业大学学报, 1996, 17(2): 23-27.
YANG G F, ZHANG X Q, LI J Q, NIE L, WANG W. Genetic diversity in Xuwen and Hainan yellow cattle based on blood protein electrophoresis. Journal of South China Agricultural University, 1996, 17(2): 23-27. (in Chinese)
Population Structure and Genetic Diversity of Lufeng Cattle and Leiqiong Cattle Based on Genome-Wide SNPs
TONG Xiong, LUO Wei, MIN Li, ZHANG ZhiFei, MA XinYan, LUO ChengLong, CHEN WeiDong, XU Bin, LI DaGang
Institute of Animal Science, Guangdong Academy of Agricultural Sciences/State Key Laboratory of Livestock and Poultry Breeding/ Heyuan Branch of Guangdong Laboratory for Lingnan Modern Agriculture/Guangdong Key Laboratory of Animal Breeding and Nutrition, Guangzhou 510640
【Objective】Phylogenetic relationship among Lufeng cattle, Leiqiong cattle and domestic cattle in different regions worldwide, and genetic diversity of different domestic cattle populations were studied, so as to lay a theoretical foundation for the identification and protection of domestic cattle resources. 【Method】Tissue samples of 12 individuals of Lufeng cattle and 17 ones of Leiqiong cattle were collected for whole genome resequencing. Combined with another 92 cattle genomes from 24 breeds worldwide available in the NCBI database, a panel of cattle genomes comprising 121 individuals were generated from 25 breeds to carry out population genetics study. Bos taurus ARS-UCD1.2 assembly was selected as the reference genome. High-quality reads were obtained by genome alignment and quality control. Genomic SNPs were detected by GATK software. Population structure was analyzed by phylogenetic tree construction, PCA clustering, and Admixture evaluation. Genetic diversity of the populations was studied by estimating nucleotide diversity (), heterozygosity (), and linkage disequilibrium (LD). 【Result】A total of 6 905 944 306 clean reads were obtained by genome sequencing from 29 individuals of the two cattle breeds in Guangdong. Average genome coverage and average sequencing depth of each sample were 97.99% and 12.78×, respectively. After integrating the NCBI cattle genome data, 14 664 391 population SNPs were identified. The results of phylogenetic tree, PCA and Admixture showed that a primary division was found betweencattle from taurine and indicine. Moreover, indicine cattle split on Chinese and Indian cattle, and Northeast Asian cattle (Hanwoo and Yanbian) and Tibetan cattle separated from European taurine group, while Wenling cattle and Zhoushan cattle differentiated from Chinese indicine group. Lufeng cattle and Leiqiong cattle belonged to pure Chinese indicine cattle. Lufeng cattle and Wannan cattle, Leiqiong cattle and Ji'an cattle showed the closest relationship, respectively. The relationships indicated that Lufeng cattle and Leiqiong cattle in the adjacent areas belonged to two independent breeds. Some Lufeng and Leiqiong individuals were genetically admixed with European taurine and Northeast Asian taurine cattle, and the admixed proportion was high, indicating that these two breeds needed to strengthen the purification and rejuvenation within the population. Compared with European taurine cattle and Korean cattle, for Chinese domestic cattle, LD decay rate was faster, and nucleotide diversity () and heterozygosity () were higher, indicating that genetic diversity of Chinese domestic cattle was richer. Compared with other Chinese domestic cattle, LD levels of Lufeng cattle and Leiqiong cattle were lower, heterozygosity () was higher, and the density distribution of nucleotide diversity () and heterozygosity () was more concentrated, indicating that the two cattle breeds were less subject to artificial selection and maintain higher genetic diversity. 【Conclusion】Population structure and genetic diversity of Lufeng cattle and Leiqiong cattle were analyzed by genome-wide SNPs, which provided data support for independent classification and conservation of these two cattle breeds.
Lufeng cattle; Leiqiong cattle; whole-genome SNPs; population structure; genetic diversity

10.3864/j.issn.0578-1752.2023.14.014
2023-01-04;
2023-04-23
广东省现代农业产业技术体系创新团队项目(2022KJ114)、岭南现代农业科学与技术广东省实验室河源分中心自主科研项目(DT20220023)、2022年省级乡村振兴战略专项资金种业振兴项目(2022-XBH-00-008)
童雄,E-mail:tongxiong@gdaas.cn。罗威,E-mail:luowei@gdaas.cn。童雄和罗威为同等贡献作者。通信作者徐斌,E-mail:xubin@gdaas.cn。通信作者李大刚,E-mail:lidagang@gdaas.cn
(责任编辑 林鉴非)
