大豆红色种皮的色素鉴定和基因定位
2023-08-15曹杰谷勇哲洪慧龙吴海涛张霞孙建强包立高邱丽娟
曹杰,谷勇哲,洪慧龙,吴海涛,张霞,孙建强,包立高,邱丽娟
大豆红色种皮的色素鉴定和基因定位
曹杰1,2,谷勇哲2,洪慧龙2,吴海涛2,张霞2,孙建强3,包立高4,邱丽娟1,2
1吉林农业大学生命科学学院,长春 130118;2中国农业科学院作物科学研究所,北京 100081;3东北农业大学农学院,哈尔滨 150030;4内蒙古自治区农牧业技术推广中心,呼和浩特 010018
【目的】揭示种子发育过程中种皮花青素(anthocyanin)的含量变化以及导致泰兴矮脚红(TXAJH)红色种皮的主要花青素成分;定位控制花青素合成积累的关键基因,为深入了解红色种皮形成的调控机制奠定基础。【方法】利用超高效液相色谱串联质谱(ultra-high performance liquid chromatography-tandem mass spectrometry,UPLC-ESI-MS/MS)检测黄色种皮大豆绥农14(SN14)和红色种皮大豆TXAJH不同发育阶段种皮的花青素成分与含量,分析与种皮颜色变化密切相关的花青素成分;利用SN14和TXAJH杂交构建的重组自交系(recombinant inbred lines,RIL)群体进行分离群体分组混合分析(bulked segregant analysis,BSA),初步定位红色种皮相关基因的候选区域,在此基础上,结合标记连锁分析缩小候选区间并预测红色种皮候选基因;最后通过qRT-PCR验证候选基因的表达情况。【结果】检测SN14和TXAJH 4个发育阶段的种皮,共发现12种花青素。在成分上,总花青素的聚类分析表明,TXAJH与SN14之间以及TXAJH显色前后之间的种皮花青素组成均存在明显差异。在含量上,种子发育过程中,SN14种皮花青素的含量逐渐下降,而TXAJH种皮的含量迅速升高并保持稳定,种皮显色后,二者的花青素含量呈现极显著差异,在成熟阶段,TXAJH种皮花青素的含量是SN14的200倍以上。矢车菊素-3-O-葡萄糖苷(Cyanidin-3-O-glucoside,Cy-3-glu)、芍药花素-3-O-葡萄糖苷(Peonidin-3-O-glucoside,Pn-3-glu)和牵牛花素-3-O-葡萄糖苷(Petunidin-3-O-glucoside,Pt-3-glu)是导致TXAJH种皮呈现红色的重要原因。BSA-seq关联分析将红色种皮基因的候选区间定位于第8染色体上,长度为8.66 Mb。利用27个多态性标记进行连锁分析得到10种单倍型,最终将候选区间缩小至702 kb。该区间中在亲本间存在非同义变异的基因共37个,其中,编码MYB转录因子,和编码bHLH转录因子,它们可能参与花青素的生物合成调控;编码花青素还原酶1,可以将花青素转化为原花青素(proanthocyanidin,PA)。基因表达分析结果表明,候选基因和花青素生物合成途径相关基因在SN14与TXAJH中的表达模式相似,均为前者低于后者。种皮花青素主要成分与候选基因表达水平的关联分析结果显示二者之间存在极强的相关性。【结论】SN14与TXAJH的种皮花青素组成存在差异,TXAJH红色种皮呈现红色可能是Cy-3-glu、Pn-3-glu和Pt-3-glu积累的结果。预测、、和为红色种皮候选基因,其中、和可能对花青素生物合成途径的多个基因产生调控作用。
大豆;种皮色;花青素;BSA-seq;基因定位;转录因子
0 引言
【研究意义】大豆((L.) Merr.)是重要的经济作物,可以为人类和牲畜提供优质的植物蛋白和油脂[1-2]。大豆种皮颜色是一类非常便于观察的生物学性状,在遗传学研究中常作为形态标记[3]。使用不同颜色来标记区分用于制药、食品饲料加工等商业用途的大豆将有望极大地降低原料筛选成本[4]。植物种皮的颜色主要取决于所合成花青素(anthocyanin)的种类和含量[5]。花青素是具有良好抗氧化活性的天然色素,长期食用花青素或富含花青素的食物对健康有一系列的好处,包括但不限于保护心血管、保护神经、改善视力、促进新陈代谢、抗菌抗炎症以及预防和治疗癌症等[6-12]。此外,花青素可以有效清除植物中由于非生物胁迫而产生的过量活性氧(reactive oxygen species,ROS),降低干旱、低温、高盐和重金属污染等逆境对植物造成的直接或间接损伤[13-17]。大豆中红色种皮较为罕见,鉴定其中的花青素成分,发掘与红色种皮形成相关基因,对解析种皮颜色形成机制,提升大豆品质和经济效益具有一定的参考意义。【前人研究进展】经典遗传研究表明,大豆籽粒颜色是一个受多位点控制的复杂性状,已发现至少9个位点(、、、、、、、和)参与控制大豆种皮颜色[18]。、和位点通过影响叶绿素代谢,使种皮表现绿色[3, 19]。、、、、和位点通过控制大豆种皮中花青素的合成与分布使种皮的整体或局部发生色素沉积进而形成各种颜色的种皮[20-28]。花青素是类黄酮(flavonoids)物质下面的重要子类,在可见光波段具有广泛的吸收范围,是使植物产生颜色的重要物质,通常导致种皮呈现棕色、红色、双色、黑色等颜色[5]。在影响大豆种皮颜色的遗传位点中,是查尔酮合成酶(chalcone synthase,CHS)基因的重复区域[20]。CHS是催化花青素生物合成途径初始步骤的专用酶,多个的存在会降低其自身在种皮中的表达水平进而抑制种皮的花青素合成[21]。和位点分别与黄烷酮3′-羟化酶(flavonoid 3′-hydroxylase,F3′H)基因和黄烷酮3′5′-羟化酶(flavonoid 3′5′-hydroxylase,F3′5′H)基因共分离[22-24]。F3′H和F3′5′H对黄烷酮B环不同位置的羟基化修饰将决定最终合成花青素的颜色类型,对植物颜色的呈现有重要影响[25-26]。和位点分别与编码R2R3-MYB转录因子和Argonaute5(AGO5)蛋白的基因有关[27-28]。R2R3-MYB转录因子调控花青素合成途径部分关键基因的表达,可导致纯黑/纯褐色种皮或双色条纹种皮[27]。AGO5蛋白具有调控siRNAs在种皮中分布的功能,使种皮特定部位例如种脐和鞍区发生色素沉积形成鞍挂种皮[28]。位点与红棕色种皮有关,但只在和位点均为隐性的时候才影响种皮颜色[18]。在基因定位方法上,传统的QTL图谱通常涉及使用分布于整个基因组中的分子标记对群体中的大量个体进行基因型和表型分析以保证足够的统计能力,这一过程需要耗费大量的时间和人力物力成本[29]。相比之下,利用分离体分组混合分析(bulked segregant analysis,BSA)方法可以通过构建目标性状极端差异的基因混池快速筛选与目标性状紧密相关的位点来定位基因[30-31]。新一代测序技术的发展迭代使更多物种陆续完成全基因组测序,基于全基因组测序的BSA方法可以在没有遗传图谱的情况下,对目标性状进行快速精准的定位分析,具有经济、方便、快捷的优点[32-33]。目前,BSA-seq方法已被广泛应用于小麦[34]、水稻[35]、玉米[36]、花生[33, 37]、大豆等的基因定位中,并取得显著成果。【本研究切入点】目前,大豆种皮花青素的研究多集中于黑色种皮的成熟种子,而对红色种皮以及种皮发育过程花青素含量变化的研究较为罕见,且已发现的位点/基因尚不能完全解释种皮颜色的形成机制。【拟解决的关键问题】本研究通过对红、黄2种颜色大豆不同发育阶段的种皮进行花青素成分含量检测,揭示种子发育过程中种皮花青素的含量变化规律以及决定红色种皮的主要花青素成分;利用绥农14与泰兴矮脚红杂交构建的重组自交系(recombinant inbred lines,RIL)群体进行BSA-seq和基因定位,预测红色种皮颜色候选基因,为深入解析种皮颜色形成的调控机制奠定基础。
1 材料与方法
1.1 植物材料
前期以黄色种皮栽培品种绥农14(SN14,ZDD22648)为母本,红色种皮地方品种泰兴矮脚红(TXAJH,ZDD04430)为父本,通过杂交和连续自交的方法构建了F9RIL群体。
SN14和TXAJH以及RIL群体的188个品系于2022年6月在中国农业科学院作物科学研究所试验田(116°20′E,39°57′N)种植,采用常规田间管理模式。播种后第30天收集亲本及每个品系的幼嫩叶片用于BSA-seq和基因定位。分别在开花后第30、40、50、60和70天剥取SN14和TXAJH的种皮用于花青素含量检测和qRT-PCR分析,每个时期的每个样品均包含3个生物重复。以上所有样品离体后立即在液氮中速冻,-80 ℃保存备用。
1.2 种皮花青素提取
分别选取SN14与TXAJH开花后第40、50、60和70天的种皮进行花青素成分检测。样品经充分研磨后,每份取0.1 g冻干粉末溶解于1 mL 70%甲醇中,充分涡旋混匀,4 °C过夜萃取,然后12 000 r/min离心10 min,吸取上清,用微孔滤膜(0.22 μm pore size)过滤得花青素提取液,避光4 ℃保存备用。
1.3 花青素检测
用超高效液相色谱串联质谱(UPLC-ESI-MS/MS)进行花青素检测。将所有待测样品的等量混合物作为QC样本,每8个待测样本中插入一个QC样本以监测在相同的处理方法下分析过程的重复性。液相条件主要包括:1)色谱柱:Waters Acquity UPLC HSS T3 C18 1.8 µm,2.1 mm×100 mm;2)流动相:各含0.1%甲酸的超纯水(A相)和乙腈(B相);3)洗脱梯度(v/v):0 min水/乙腈为95/5,10.0 min为5/95,11.0 min为5/95,11.1 min为95/5,15.0 min为95/5;4)流速0.4 mL·min-1;柱温40 ℃;进样量2 μL。质谱条件主要包括:电喷雾离子源(electrospray ionization,ESI)温度为550 ℃,质谱电压为5 500 V/-4 500 V,离子源气体Ⅰ(gasⅠ,GSⅠ)为55 psi,气体Ⅱ(gasⅡ,GSⅡ)为60 psi,气帘气(curtain gas,CUR)为25 psi,碰撞诱导电离(collision-activated dissociation,CAD)参数设置为高。检测结束后,根据二级谱信息利用岛津株式会社(https://www.shimadzu.com/)提供的化合物比对数据库进行花青素定性。以二甲基亚砜(dimethyl sulfoxide,DMSO)为标准品,利用三重四级杆质谱的多反应监测模式(multiple reaction monitoring,MRM)进行花青素的定量分析。
1.4 DNA和RNA提取
将保存于-80 ℃的叶片和种皮样品分别在液氮中充分研磨。每份叶片样本取0.1 g冻干粉末,用CTAB法提取基因组DNA[38]。每份种皮样本取0.1 g冻干粉末,用植物总RNA提取试剂盒FastPure®Universal Plant Total RNA Isolation Kit(#RC411, Vazyme)按照产品说明书提取种皮总RNA。用Gene Company Limited(基因有限公司)的NanoDrop-1000分光光度计检测DNA和RNA的浓度和质量,-80 ℃保存备用。
1.5 BSA测序分析
1.5.1 文库构建和测序 依据《大豆种质资源描述规范和数据标准》[39],从RIL群体中选择30个黄色种皮品系和30个红色种皮品系,将DNA分别等量混合,构成2个子代基因池,命名为Y和R。在亲本SN14和TXAJH中各取10个单株,将DNA分别等量混合,构成2个亲本基因池,命名为SN和TX。
文库的构建和测序由北京百迈克生物科技有限公司完成。试验流程按照Illumina公司提供的标准protocol执行,首先用超声破碎的方法将DNA随机打断成350 bp大小的片段,DNA片段经末端修复、3′端加A、加测序接头、纯化后进行PCR扩增,最终通过Illumina HiSeq进行测序。亲本池测序深度为10×,后代混池测序深度为30×。
1.5.2 变异检测和关联分析 利用Bcltofastq(v1.8.4)对测序结果进行碱基识别,原始序列过滤得到clean reads,以大豆参考基因组(Wm82.a2.v1)为模板,利用bwa软件对clean reads进行拼接组装[40]。使用samtools(v1.9)过滤冗余reads。使用GATK的HaplotypeCaller(局部单体型组装)算法进行SNP和InDel的变异检测[41-42]。使用SnpEff软件对变异位点集进行注释和预测[43]。通过欧式距离(euclidean distance,ED)算法和index算法进行关联分析[44-45]。ED算法用于计算变异位点与性状的关联程度,ED值越大表明该位点在两混池间的差异越大。ED值计算公式如下:
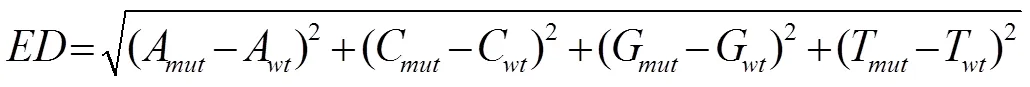
式中,A、C、G、T分别表示碱基A、C、G、T在突变混池中的频率,A、C、G、T分别表示碱基A、C、G、T在野生型混池中的频率。
index算法用于寻找混池之间变异位点基因型频率显著差异的位点,变异位点与性状关联度越强,则∆index越接近于1。index值计算方法如下:
()=/(+)
()=/(+)
Δ=()-()
式中,()表示()池来源于母本的深度,()表示()池来源于父本的深度。
对关联分析结果区域应用BLAST软件在NR[46]、Swiss-Prot、GO[47]、COG[48]、KEGG[49]等数据库中对候选区域编码基因进行匹配和注释。
1.6 基因定位
利用Misa软件在BSA候选区域检索SSR位点,通过SoyBase(https://soybase.org/)网站从大豆参考基因组(Wm82.a2.v1)提取位点上下游200 bp序列,使用NCBI网站(https://www.ncbi.nlm.nih.gov/tools/ primer-blast/)进行引物设计(电子附表1)。将DNA用ddH2O稀释至50 ng·µL-1后用于PCR反应。使用96孔板配置50 µL反应体系,包括25 µL 2×Rapid Taq Master Mix(#P222,Vazyme)、1 µL稀释后的DNA、各2 µL的正向和反向引物(10 µmol·L-1),以及20 µL ddH2O。在BIO-RED T100 PCR仪上进行反应,反应程序为95 ℃ 3 min;95 ℃ 15 s,57 ℃ 15 s,72 ℃ 15 s,35个循环;72 ℃ 5 min。PCR产物经6%聚丙烯酰胺凝胶电泳法分离,银染后读取基因型。
1.7 qRT-PCR分析
使用NCBI(https://www.ncbi.nlm.nih.gov/tools/ primer-blast/)网站设计引物(电子附表2)。每个样品取1 000 ng总RNA使用逆转录试剂盒HiScript®III RT SuperMix for qPCR(+gDNA wiper,#R323,Vazyme)进行逆转录,产物经无核酸酶水稀释至1 ng·µL-1后用于qRT-PCR分析。使用Hard-Shell®384孔板(BIO-RAD)配置20 µL反应体系10 µL Taq Pro Universal SYBR qPCR Master Mix(#Q712,Vazyme)、5 µL稀释后的cDNA、各2 µL的正向和反向引物(10 µmol·L-1),以及4.2 µL无核酸酶水。在QuantStudio 7 Flex上进行反应。反应程序为95 ℃ 30 s;95 ℃ 10 s,60 ℃ 30 s,40个循环;熔解曲线分析为95 ℃ 10 s,60 ℃ 60 s,95 ℃ 15 s。以无模板反应作为阴性对照,每个反应设置3个技术重复。
1.8 数据分析
利用卡方分析检验群体的分离比例,计算方法如下:
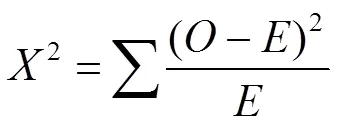
式中,表示实际频数,表示理论频数。
使用作为内参基因,根据2-∆∆Ct方法计算qRT-PCR反应中目的基因的相对表达量,采用log2计算以后的FPKM值绘制热图。用GraphPad Prism 9(v9.4.1)软件进行统计分析和绘图。
2 结果
2.1 种皮花青素含量差异
2.1.1 不同发育阶段的种皮颜色分析 从开花后第30天开始对SN14和TXAJH的种皮颜色进行以10 d为间隔的连续观察直至种子脱水成熟(第70天)。如图1所示,在第40天以前,二者种皮均为绿色,从第50天开始,SN14种皮逐渐变黄而TXAJH种皮则出现红色且逐渐加深,表明导致二者种皮颜色差异的代谢物在第40天以后才开始出现明显积累。

图1 SN14和TXAJH不同时期种皮颜色
2.1.2 种皮着色过程花青素含量分析 为了解种皮发育过程中花青素的含量变化,利用UPLC-ESI- MS/MS方法检测了SN14和TXAJH在开花后第40、50、60和70天的种皮花青素成分。对SN14和TXAJH各时期种皮样本的总花青素进行层次聚类分析,以了解各组样本之间花青素的总体差异和组内样本之间的重复性。如图2-A所示,花青素总成分的聚类分析将SN14与TXAJH分别聚为一类,TXAJH显色前后的种皮也分别聚为一类。各组样本间的重复性良好,但SN14第60天的1个生物重复样本(S_60_2)与第50天的样本(S_50_1,2,3)聚到一起,表明前者在组间的相似性高于组内,因此,将S_60_2样本剔除再进行后续分析。
在SN14和TXAJH各时期的种皮中共检测到12种花青素。如图2-B所示,在种子成熟过程中花青素总含量随种皮颜色变化而变化。TXAJH种皮内花青素含量在着色前较低,第50天出现激增并继续升高,在第60天以后含量稳定;而SN14种皮内花青素含量较低,且随着种子的成熟持续下降。这一趋势与SN14和TXAJH种皮颜色变化情况一致,表明种皮颜色与种皮花青素含量相关。在被检测的4个时期中,TXAJH种皮的色素含量始终高于SN14,种皮显色后这一差异达到极显著水平。红色时期的TXAJH种皮中含量占比最高的3种花青素分别是矢车菊素-3-O-葡萄糖苷(Cyanidin-3-O-glucoside,Cy-3-glu)、芍药花素-3-O-葡萄糖苷(Peonidin-3-O-glucoside,Pn-3-glu)和牵牛花素-3-O-葡萄糖苷(Petunidin-3-O-glucoside,Pt-3- glu);而SN14则分别为矢车菊素(Cyanidin,Cy)、天竺葵素-3-O-葡萄糖苷(Pelargonidin-3-O-beta-D- glucoside,Pg-3-glu)和锦葵色素-3-O-葡萄糖苷(Malvidin-3-O-glucoside,Mv-3-glu)。2组花青素的总含量在成熟种子的种皮中相差超过200倍。由此推测Cy-3-glu、Pn-3-glu和Pt-3-glu 3种花青素是导致TXAJH种皮显示红色的重要原因。
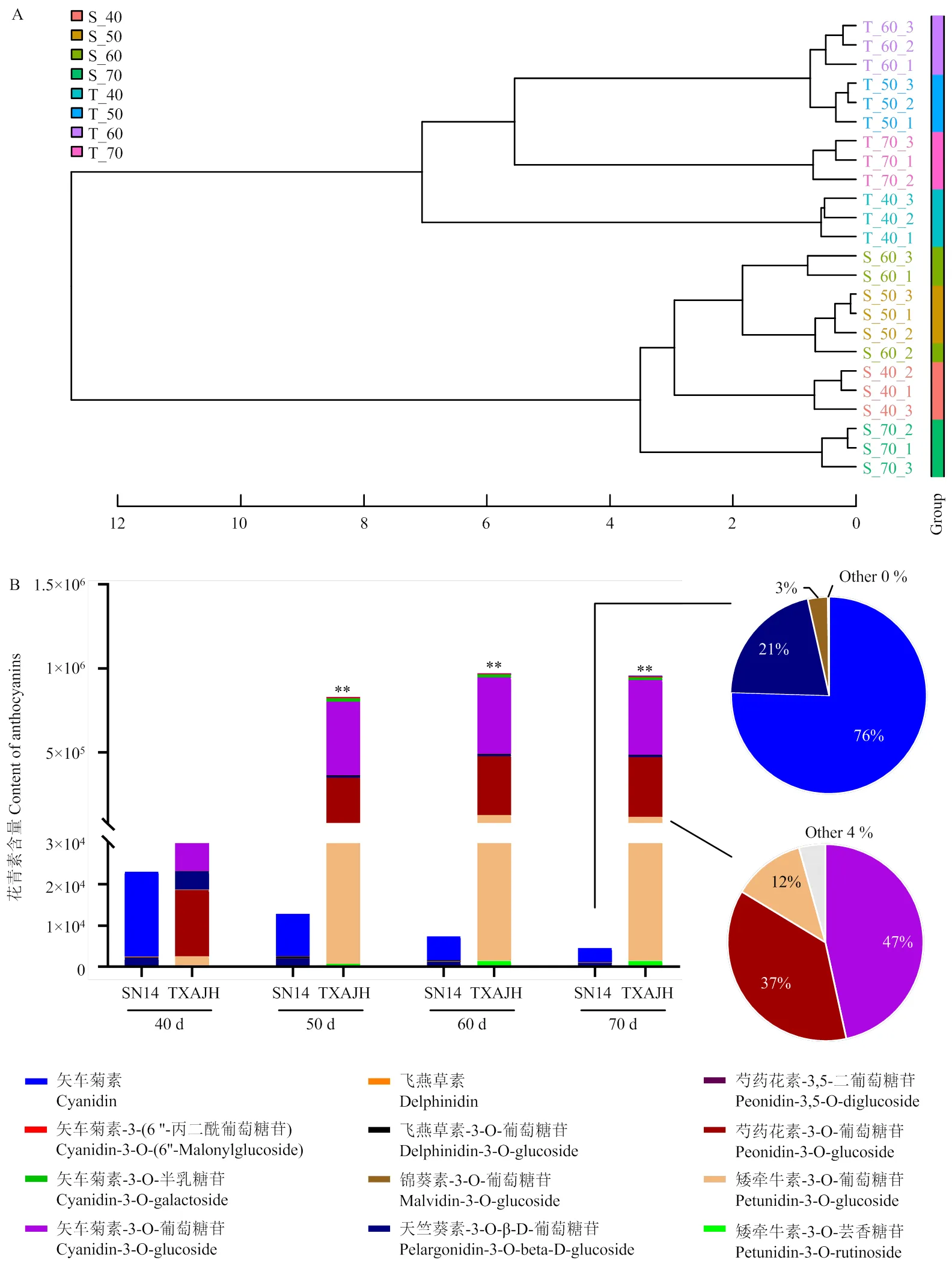
A:种皮总花青素聚类分析,横坐标为不同样本之间的距离,纵坐标为样本编号,其中,S:SN14;T:TXAJH,字母后数字分别表示相应时期和生物重复,不同样本的组别通过颜色区分。B:种皮发育过程花青素含量分析,柱形图表示花青素含量,饼图表示70 d时花青素在SN14和TXAJH中的相对占比,同一花青素用相同颜色填充,**表示差异极显著(P<0.01)
2.2 BSA-seq初步定位候选区间
2.2.1 种皮色性状的遗传分析 前期以黄色种皮品种SN14和红色种皮品种TXAJH为亲本通过杂交和单粒下传的方式最终构建了F9RIL群体。其中,F1种皮颜色为黄色,表明种皮颜色性状黄色相对红色为显性。在F9RIL群体中,黄色种皮的品系共109个,红色种皮的品系共79个。如表1所示,卡方值2= 4.556>2(0.05, 1)=3.841,表明种皮颜色性状分离比不符合预期的3﹕1,即红色种皮基因受2对及以上的等位基因控制。

表1 种皮色性状遗传分析
2.2.2 数据质控和变异位点检测 对Illumina HiSeq测序得到的原始结果进行数据过滤,主要过滤低质量和不可信的reads。过滤后共得到110.22 Gb的清洁数据(clean read),Q30平均达到93.26%,样品与参考基因组平均可比对率为99.22%,基因组平均覆盖深度为23.5×,基因组碱基被至少覆盖1次的平均比例为98.44%。以上结果表明测序随机性良好,可用于后续分析。亲本及混池样品测序数据统计如表2所示。
根据Clean Reads的定位结果,剔除重复Reads后用GATK进行SNP和InDel的变异检测,每个样本先各自生成gVCF,再进行群体joint-genotype。最后过滤掉低可信度或重复的位点得到最终的变异位点集,通过对各样品间的SNP和InDel进行Venn统计(图3),在具有相同种皮颜色的亲本和混池中同时存在的位点可能与相应种皮颜色的形成有关。统计结果显示,共有2 285 504个SNP变异位点和501 857个InDel变异位点可能与种皮颜色相关。

SNP_veen:样品间SNP位点统计结果;InDel_veen:样品间InDel位点统计结果。变异位点的统计只涉及位点的位置而与基因型无关
2.2.3 关联分析 通过对得到的变异位点集进行关联分析以获得相关基因的候选区域(图4),SNP-ED关联分析得到1个候选区域,包含1 174个基因;SNP-index关联分析得到3个携带基因的候选区域,共包含1 141个基因;InDel-ED关联分析得到1个携带基因的候选区域,包含1 201个基因;InDel-index关联分析得到1个携带基因的候选区域,包含1 132个基因。所有候选区域的相关信息详见电子附表3。
综上,对不同关联分析方法得到的SNP和InDel关联区域结果取交集(表3),最终得到1个候选区域,将该区域作为红色种皮基因候选区间。
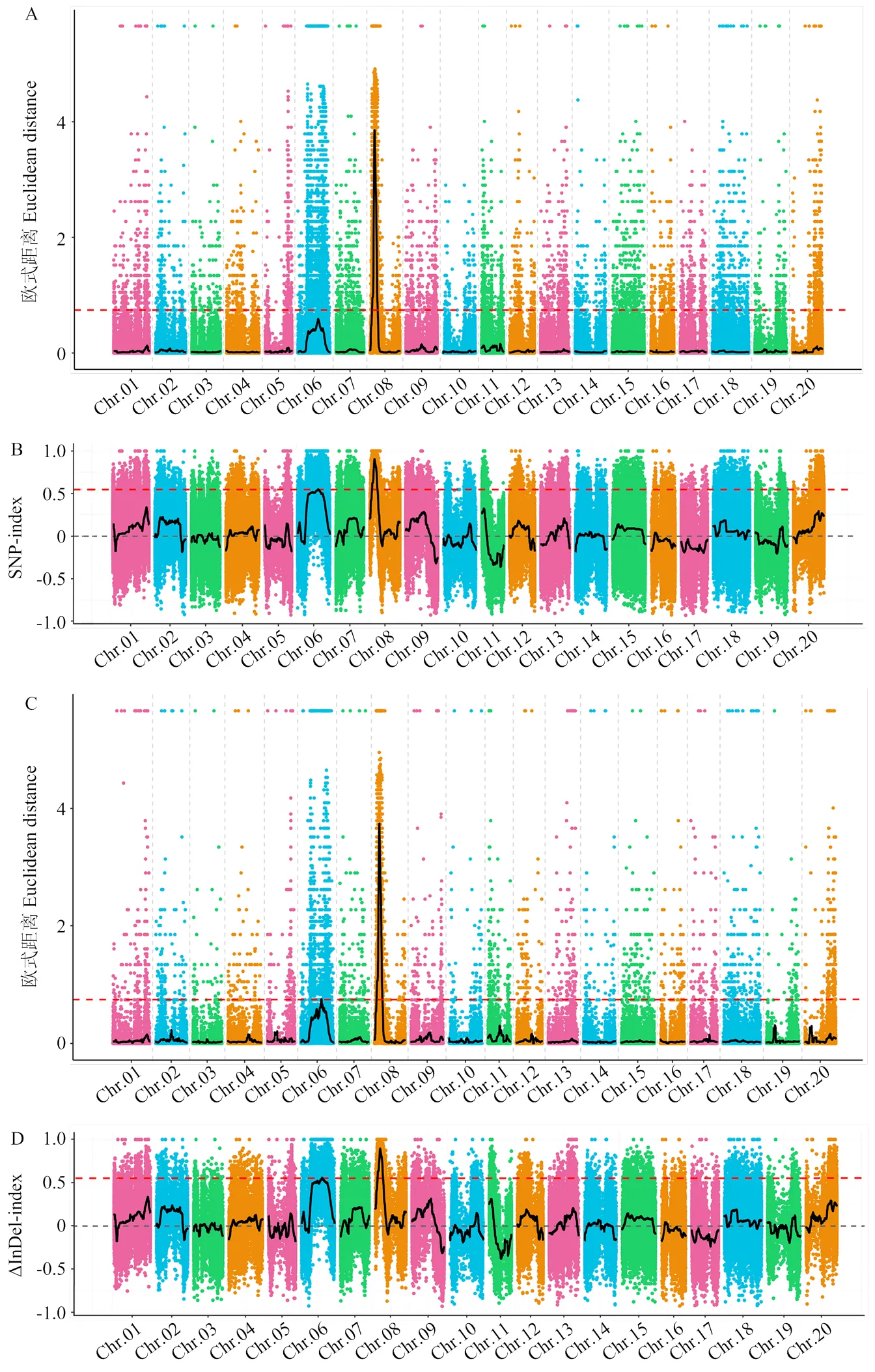
A:SNP-ED关联结果;B:SNP-index关联分析的结果;C:InDel-ED关联结果;D:InDel-index关联分析的结果。横坐标为染色体名称,彩色的点代表每个SNP(或InDel)位点的ED(或Δindex)值,黑色线为拟合后的ED(或Δindex)值,红色虚线代表显著性关联阈值
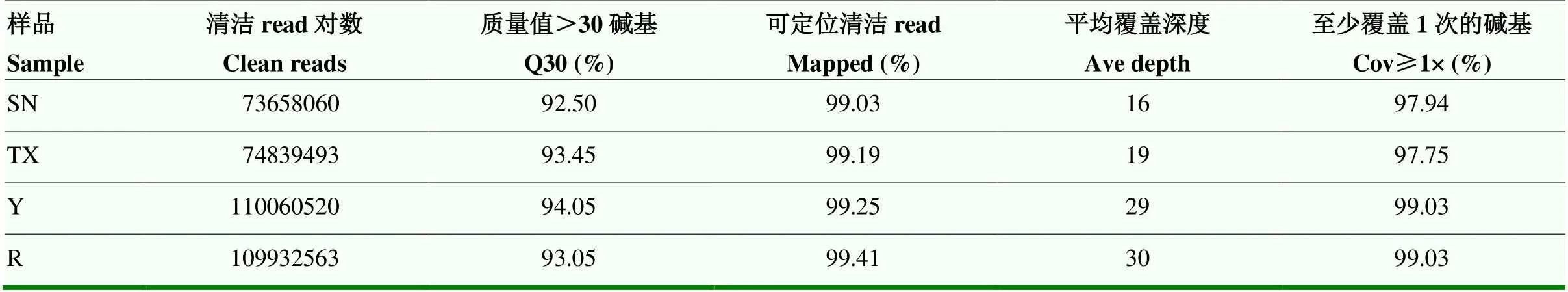
表2 样品测序数据统计

表3 不同关联分析方法得到的共同候选区域
2.3 标记连锁分析和候选基因预测
2.3.1 标记连锁分析 为缩小大豆红色种皮候选基因的范围,基于BSA候选区域筛选了27对在亲本及子代基因池之间具有多态性的SSR引物(电子附表1)对RIL群体中188个品系(红色种皮品系79个,黄色种皮品系109个)进行标记-表型连锁分析。共获得10种单倍型(图5),其中4种来自于红色种皮(Red),另6种来自于黄色种皮(Yellow),将候选区间缩小至标记08-223与08-259之间,长度约702 kb。区间内在亲本间有37个基因发生非同义变异。
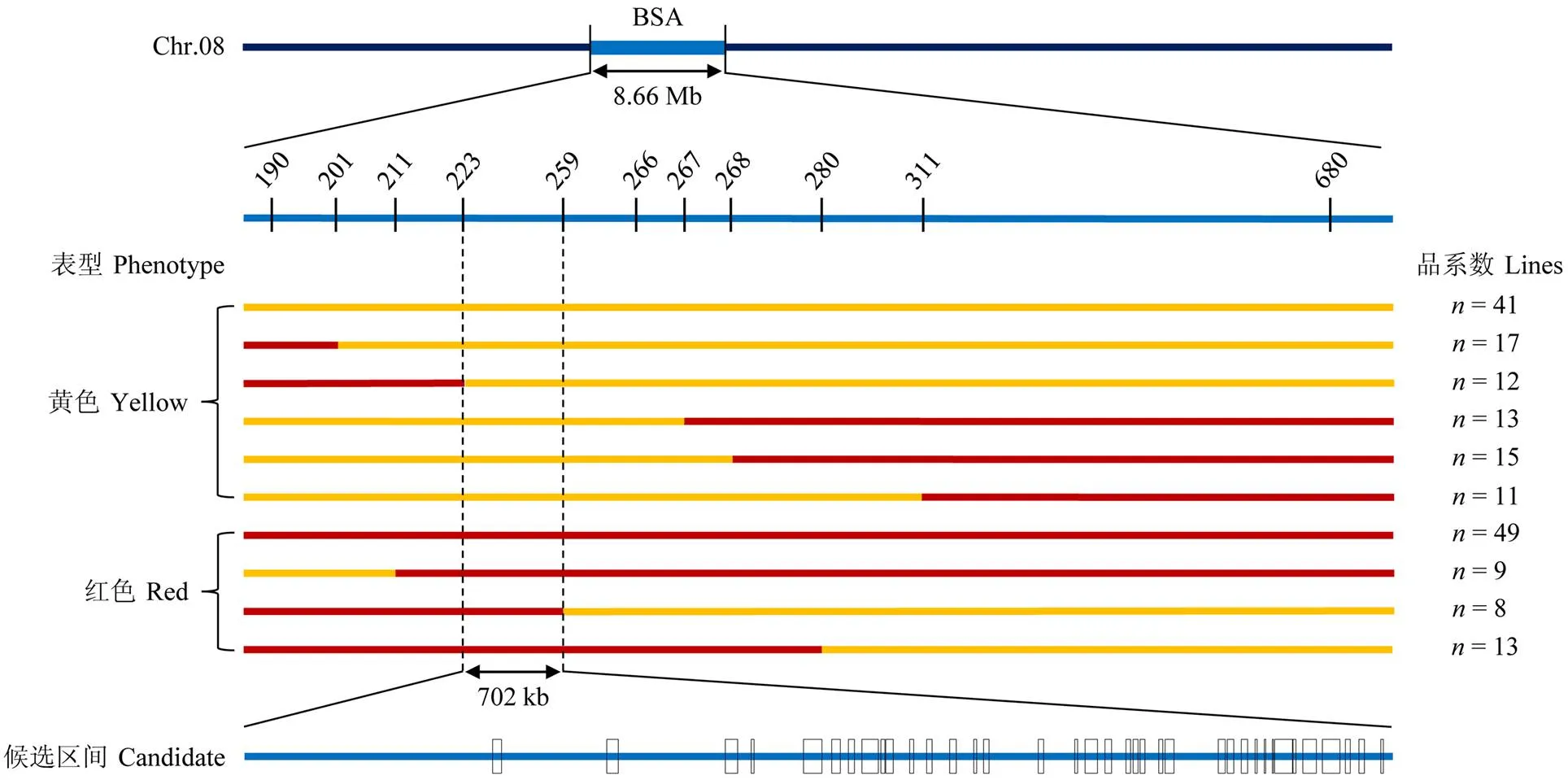
图5 种皮颜色候选区间标记连锁分析
2.3.2 候选基因预测 在Phytozome(https://phytozome- next.jgi.doe.gov/)网站中对定位区间内的37个基因进行功能注释(表4),、、和在数据库中功能未知,推测可能为假基因或大豆特有新基因。在成功注释的33个基因中,为MYB转录因子基因,和为bHLH转录因子基因,它们可以调控花青素生物合成途径相关基因的表达;为花青素还原酶1基因,催化花青素向原花青素的转化。花青素是影响植物种皮颜色的重要物质,因此,预测这4个基因可能为大豆红色种皮候选基因。
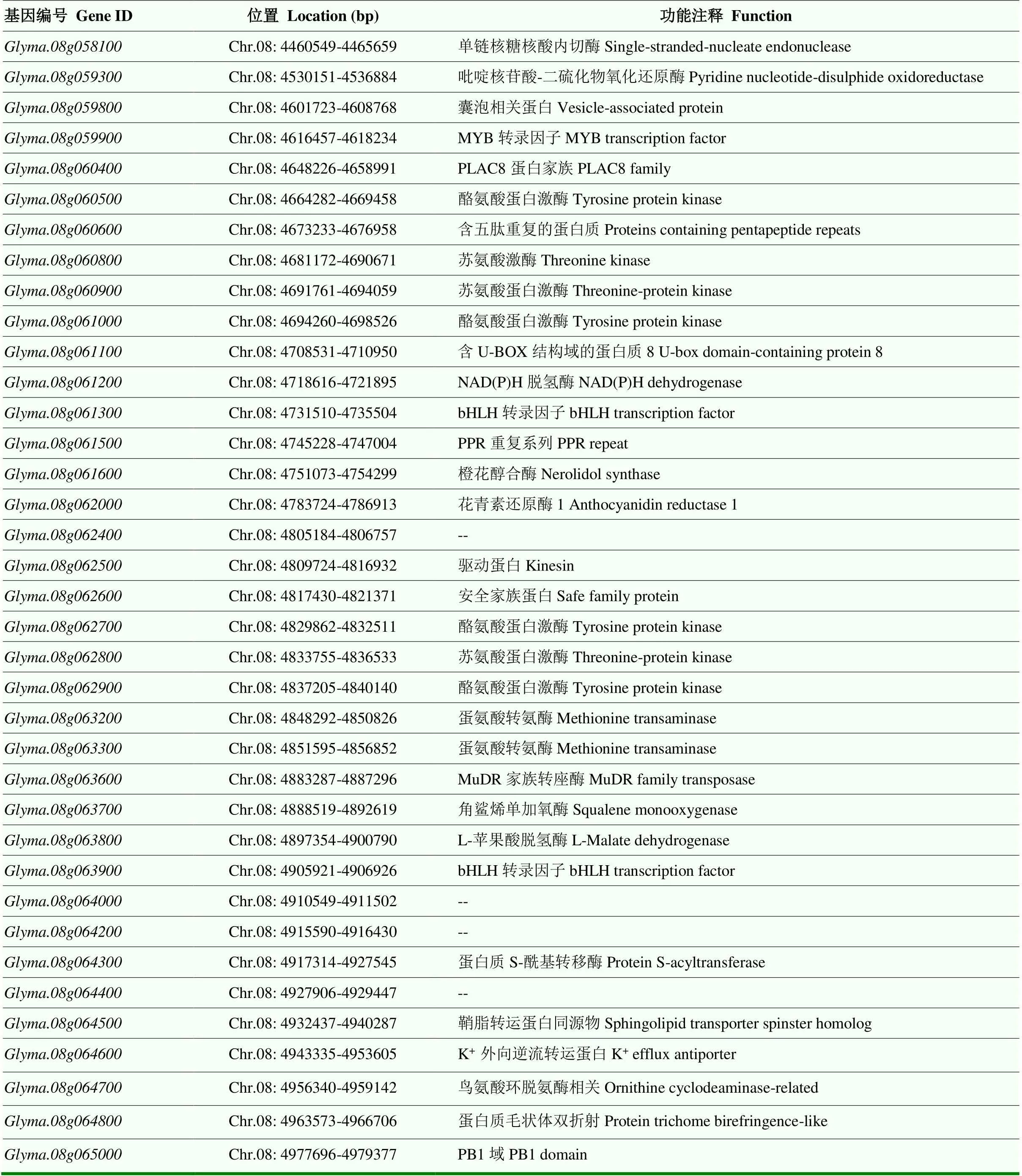
表4 候选区间基因及功能注释
2.4 基因表达分析
为探究候选基因在种皮发育过程中的表达模式,用SN14和TXAJH在开花后第30、40、50、60和70天的种皮总RNA进行qRT-PCR分析(图6),在种皮发育的5个时期,候选基因在SN14与TXAJH中的表达模式存在区别。在SN14种皮发育的各时期表达水平较低,仅在发育前期出现相对较高的表达水平,但仍低于同时期的TXAJH;在TXAJH种皮中,各候选基因的表达水平均较高,且与种皮的花青素含量之间具有相似的变化趋势。将SN14和TXAJH种皮发育过程中6种主要花青素的含量与候选基因的表达水平进行关联分析(图6-B),结果表明,它们之间存在相关性。其中,Cy和Mv-3-glu的积累与4个候选基因的表达水平之间存在较明显的负相关;和的表达水平与Cy-3-glu、Pt-3-glu、Pg-3-glu和Pn-3-glu的积累之间存在正相关且具有显著差异。此外,定位区间内其他33个基因的qRT-PCR结果显示,它们在SN14与TXAJH种皮中的表达模式没有明显区别,表明相关基因与种皮花青素积累之间的相关性较低。
通过查阅文献[50]选择了8个花青素生物合成途径的结构基因进行表达量分析(图6-C),在绝大多数时期,TXAJH种皮中这些基因的表达水平均显著高于SN14。这表明候选基因、和可能在花青素生物合成途径中不仅局限于对某一个结构基因产生调控,而是对通路中多个基因的表达都具有调控作用。
3 讨论
3.1 红色种皮的形成与Cy-3-glu、Pn-3-glu和Pg-3- glu的积累有关
大豆有着悠久的种植历史,在漫长的驯化与选育过程中形成了丰富多样的种皮颜色[51]。在多种豆科作物中种皮颜色都被证实与其内的多酚类物质含量相关[52]。大豆也不例外,Malenčić等[53]发现大豆种皮颜色的深浅与种皮花青素(属多酚类)的含量有关。然而,现有大豆种皮花青素的研究多集中于成熟种子,对种子发育过程中种皮花青素含量的研究较为罕见。本研究通过检测SN14和TXAJH在4个发育阶段种皮的花青素含量,发现种皮花青素含量随着种皮颜色的加深而升高。在种皮发育过程中,TXAJH的种皮花青素含量始终高于SN14,有趣的是这些差异在绿色种皮阶段并不显著,而从第50天开始变得极显著。Cy-3-glu、Pn-3-glu和Pg-3-glu是TXAJH种皮花青素中含量占比最高的3种色素,在SN14与TXAJH种皮颜色出现差异后,三者在TXAJH种皮中的含量显著增加,与此同时它们在SN14种皮中未检出。结果表明,TXAJH的红色种皮可能主要是这三种花青素逐步积累的结果。
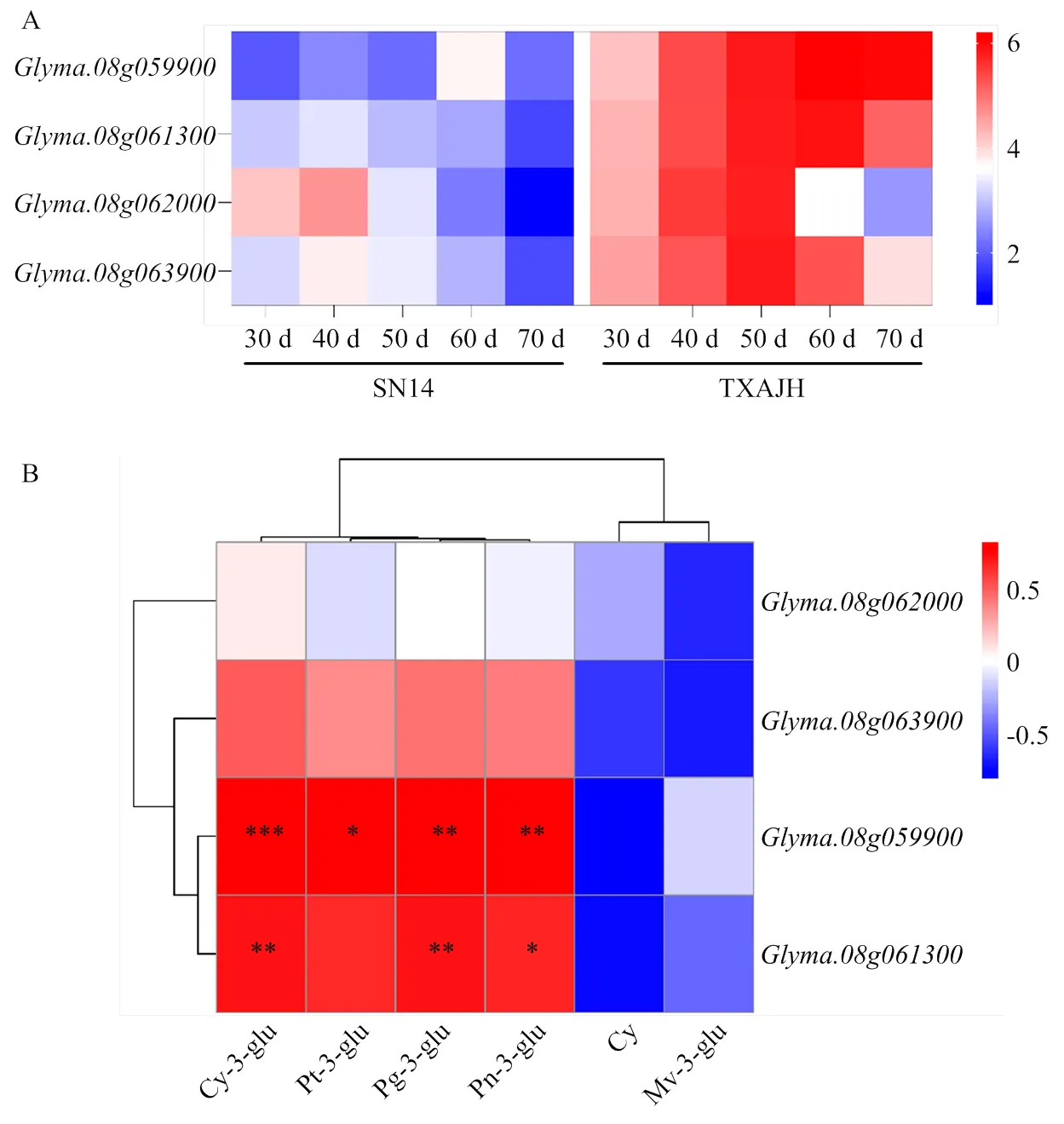
A:候选基因表达热图,采用log2计算以后的FPKM值进行作图,颜色梯度可视化基因相对表达水平;B:候选基因与种皮主要花青素间的关联热图,横坐标为花青素,纵坐标为基因,颜色梯度可视化花青素与基因间的相关性系数;C:花青素合成途径相关基因在种皮各时期的表达情况。*:<0.05,**:<0.01,***:<0.001
A: Heat map of candidate gene expression, plotted using FPKM values after Log2calculations, with color gradients to visualize relative gene expression levels; B: Heat map of the association between candidate genes and the major anthocyanins of the seed coat, the horizontal coordinate is anthocyanins and the vertical coordinate is genes, the color gradient visualizing the correlation coefficient between the anthocyanins and the genes; C: Expression of genes related to the anthocyanin synthesis pathway at various times in the seed coat. *:<0.05, **:<0.01, ***:<0.001
图6 候选基因表达及关联分析
Fig. 6 Candidate gene expression and association analysis
3.2 BSA-seq快速初定位红色种皮基因候选区间
在过去较长的一段时间里,基于图位克隆方法,利用目标基因紧密连锁分子标记在染色体上的位置来逐步确定和分离目标基因是许多物种确定候选基因的主要方法[54]。但该方法需要在全基因组范围大规模筛选标记,耗时费力,限制了相关优异基因资源的探索。本研究使用基于全基因组测序的BSA分析方法,通过变异位点与目标性状的关联分析,快速将种皮色基因候选区域定位于第8染色体(Chr.08:3 530 000—12 280 000 bp),提高了基因定位的效率。此外,测序结果提供了候选区域内基因序列的变异信息,既可以在进一步候选区间缩小中为标记开发提供依据,也可以了解变异对相关基因结构的影响,为候选基因的筛选提供依据。本研究通过BSA-seq得到的初定位区间大小为8.6 Mb,包含1 127个基因,区间长度较大,且在index关联分析结果中变异位点与性状的关联强度未超过理论阈值,推测可能是由于构建子代极端混池用的样本量偏小,不能充分消除遗传背景噪音所致。但这并不影响候选区域的准确性,这一现象在其他物种和性状的研究中曾有先例[55-56]。BSA-seq与标记连锁分析相结合是解决这一问题行之有效的办法[57]。Lei等[58]通过BSA-seq和经典QTL图谱将水稻耐盐QTL从4.17 Mb缩小至222 kb,并鉴定最终候选基因;Zhao等[37]通过BSA-seq将花生紫色种皮基因定位在4.7 Mb区间,随后结合精细定位将范围缩小至2.7 kb并最终鉴定到候选基因。因此,BSA-seq结合标记连锁分析对于定位主要QTL或挖掘目标基因是必要的。
3.3 红色种皮候选基因的预测与表达验证
花青素的生物合成调控方式在多种植物中得到了广泛的研究[59-61]。大量研究发现调节植物花青素合成的转录因子主要为MYB、bHLH和WD40,它们可以单独发挥调控作用,但主要还是以形成MBW复合物的方式共同进行调控[62]。与用途广泛的bHLH和WDR转录因子相比,特异性的MYB转录因子是决定MBW复合物靶点的关键因素[63-64],已在多种植物中分离并鉴定,如草莓[65]、胡萝卜[66]、海棠[67]和矮牵牛杂交种[68]等。迄今为止,大豆中有4个MYB转录因子基因()、()、()和()被鉴定出来与大豆花青素合成相关[26, 64, 69]。在本研究得到的4个候选基因中,曾被证明为ANR1基因[70],而、和在此前的种皮颜色研究中未曾被报道。与MYB转录因子相关,和与转录因子bHLH相关,在种皮着色的5个阶段,它们在TXAJH种皮中的表达水平均高于同时期的SN14,基因表达与主要花青素含量的关联分析表明二者之间存在极强的相关性。此外,在种皮着色的各时期,TXAJH种皮中花青素合成途径相关结构基因的表达水平均高于SN14,这一表达模式与3个转录因子相关候选基因相似,暗示着、和可能不止对某一个结构基因产生了调控作用,而是同时影响了多个基因的表达。但这一猜测还需更深入的研究证实。
4 结论
SN14和TXAJH种皮的颜色与种皮中花青素的组成和含量有关,Cy-3-O-glu、Pn-3-O-glu和Pt-3-O-glu是使TXAJH种皮显红色的重要原因。、和在种皮中的表达水平和花青素的含量之间具有极强的相关性。、和可能对花青素合成途径的多个结构基因产生了调控作用。
[1] Guo B F, Sun L P, Jiang S Q, Ren H L, Sun R J, Wei Z Y, Hong H L, Luan X Y, Wang J, Wang X B, Xu D H, Li W B, Guo C H, Qiu L J. Soybean genetic resources contributing to sustainable protein production. Theoretical and Applied Genetics, 2022, 135(11): 4095-4121.
[2] Fang C, Kong F J. Soybean. Current Biolog, 2022, 32(17): R902-R904.
[3] 宋健. 大豆种皮色相关基因的图位克隆及功能解析[D]. 北京: 中国农业科学院, 2019.
SONG J. Mapping cloning and functional analysis of genes relatead to oybean seed coat color[D]. Beijing: Chinese Academy of Agricultural Sciences, 2019. (in Chinese)
[4] Kovinich N, Saleem A, Rintoul T L, Brown D C W, Arnason J T, Miki B. Coloring genetically modified soybean grains with anthocyanins by suppression of the proanthocyanidin genesand. Transgenic Research, 2012, 21(4): 757-771.
[5] Enaru B, Drețcanu G, Pop T D, Stǎnilǎ A, Diaconeasa Z. Anthocyanins: factors affecting their stability and degradation. Antioxidants, 2021, 10(12): 1967.
[6] Pojer E, Mattivi F, Johnson D, Stockley C S. The case for anthocyanin consumption to promote human health: a review. Comprehensive Reviews in Food Science and Food Safety, 2013, 12(5): 483-508.
[7] Lee Y M, Yoon Y, Yoon H, Park H M, Song S, Yeum K J. Dietary anthocyanins against obesity and inflammation. Nutrients, 2017, 9(10): 1089.
[8] Mattioli R, Francioso A, Mosca L, Silva P. Anthocyanins: a comprehensive review of their chemical properties and health effects on cardiovascular and neurodegenerative diseases. Molecules, 2020, 25(17): 3809.
[9] Liu J Q, Zhou H B, Song L, Yang Z J, Qiu M, Wang J, Shi S L. Anthocyanins: promising natural products with diverse pharmacological activities. Molecules, 2021, 26(13): 3807.
[10] Nomi Y, Iwasaki-Kurashige K, Matsumoto H, TSUDA T, KALT W. Therapeutic effects of anthocyanins for vision and eye health. Molecules, 2019, 24(18): 3311.
[11] Ngamsamer C, Sirivarasai J, Sutjarit N. The benefits of anthocyanins against obesity-induced inflammation. Biomolecules, 2022, 12(6): 852.
[12] de Arruda Nascimento E, de Lima Coutinho L, da Silva C J, de Lima V L A G, Dos Santos Aguiar J.anticancer properties of anthocyanins: a systematic review. Biochimica et Biophysica Acta Reviews on Cancer, 2022, 1877(4): 188748.
[13] Naing A H, Kim C K. Abiotic stress-induced anthocyanins in plants: their role in tolerance to abiotic stresses. Physiologia Plantarum, 2021, 172(3): 1711-1723.
[14] Zhang Q L, Zhai J J, Shao L, Lin W, Peng C L. Accumulation of anthocyanins: an adaptation strategy ofto low temperature in winter. Frontiers in Plant Science, 2019, 10: 1049.
[15] Waseem M, Rong X Y, Li Z G. Dissecting the role of a basic-loop-transcription factor,, under salt and drought stresses in transgenicL.. Frontiers in Plant Science, 2019, 10: 734.
[16] Chunthaburee S, Sakuanrungsirikul S, Wongwarat T, Sanitchon J, Pattanagul W, Theerakulpisut T. Changes in anthocyanin content and expression of anthocyanin synthesis genes in seedlings of black glutinous rice in response to salt stress. Asian Journal of Plant Sciences, 2016, 15(3/4): 56-65.
[17] Ai T N, Naing A H, Yun B W, Lim S H, Kim C K. Overexpression ofenhances anthocyanin accumulation and heavy metal stress tolerance in transgenic. Frontiers in Plant Science, 2018, 9: 1388.
[18] 邱红梅, 陈亮, 侯云龙, 王新风, 陈健, 马晓萍, 崔正果, 张玲, 胡金海, 王跃强, 邱丽娟. 大豆种子颜色遗传调控机制研究进展. 作物学报, 2021, 47(12): 2299-2313.
Qiu H M, Chen L, Hou Y L, Wang X F, Chen J, Ma X P, Cui Z G, Zhang L, Hu J H, Wang Y Q, Qiu L J. Research progress on genetic regulatory mechanism of seed color in soybean (). Acta Agronomica Sinica, 2021, 47(12): 2299-2313. (in Chinese)
[19] Wang M, Li W Z, Fang C, Xu F, Liu Y C, Wang Z, Yang R, Zhang M, Liu S L, Lu S J, Lin T, Tang J Y, Wang Y Q, Wang H R, Lin H, Zhu B G, Chen M S, Kong F J, Liu B H, Zeng D L, Jackson S A, Chu C C, Tian Z X. Parallel selection on a dormancy gene during domestication of crops from multiple families. Nature Genetics, 2018, 50(10): 1435-1441.
[20] Cho Y B, Jones S I, Vodkin L O. Nonallelic homologous recombination events responsible for copy number variation within an RNA silencing locus. Plant Direct, 2019, 3(8): e00162.
[21] Todd J J, Vodkin L O. Duplications that suppress and deletions that restore expression from asynthase multigene family. The Plant Cell, 1996, 8(4): 687-699.
[22] Guo Y, Qiu L J. Allele-specific marker development and selection efficiencies for both flavonoid 3’- hydroxylase and flavonoid 3’,5’-hydroxylase genes in soybean subgenus. Theoretical and Applied Genetics, 2013, 126(6): 1445-1455.
[23] Zabala G, Vodkin L. A putative autonomous 20.5 kb-CACTA transposon insertion in anallele identifies a new CACTA transposon subfamily in. BMC Plant Biology, 2008, 8: 124.
[24] Zabala G, Vodkin L O. A rearrangement resulting in small tandem repeats in thegene of white flower genotypes is associated with the soybeanlocus. Crop Science, 2007, 47(S2): s113-s124.
[25] Nitarska D, Stefanini C, Haselmair-Gosch C, Miosic S, Walliser B, Mikulic-Petkovsek M, Regos I, Slatnar A, Debener T, Terefe-Ayana D, Vilperte V, Hadersdorfer J, Stich K, Halbwirth H. The rare orange-red colored Euphorbia pulcherrima cultivar ‘Harvest Orange’ shows a nonsense mutation in a flavonoid 3'-hydroxylase allele expressed in the bracts. BMC Plant Biology, 2018, 18(1): 216.
[26] Vikhorev A V, Strygina K V, Khlestkina E K. Duplicated flavonoid 3’-hydroxylase and flavonoid 3’, 5’- hydroxylase genes in barley genome. PeerJ, 2019, 7: e6266.
[27] Zabala G, Vodkin L O. Methylation affects transposition and splicing of a large CACTA transposon from a MYB transcription factor regulating anthocyanin synthase genes in soybean seed coats. PLoS One, 2014, 9(11): e111959.
[28] Cho Y B, Jones S I, Vodkin L O. Mutations inilluminate epistatic interactions of theandloci leading to saddle seed color patterns in. The Plant Cell, 2017, 29(4): 708-725.
[29] Zou C, Wang P X, Xu Y B. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnology Journal, 2016, 14(10): 1941-1955.
[30] Michelmore R W, Paran I, Kesseli R V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proceedings of the National Academy of Sciences of the United States of America, 1991, 88(21): 9828-9832.
[31] Zhang G, Angeles E R, Abenes M L P, Khush G S, Huang N. RAPD and RFLP mapping of the bacterial blight resistance gene xa-13 in rice. Theoretical and Applied Genetics, 1996, 93(1): 65-70.
[32] Ma J J, Zhao Y H, Chen H, Fu C, Zhu L, Zhou X M, Xia H, Hou L, Li G H, Zhuang W J, Wang X J, Zhao C Z. Genome-wide development of polymorphic microsatellite markers and their application in peanut breeding program. Electronic Journal of Biotechnology, 2020, 44: 25-32.
[33] Pan J W, Zhou X M, Ahmad N, Zhang K, Tang R H, Zhao H L, Jiang J, Tian M D, Li C S, Li A Q, Zhang X Y, He L Q, Ma J, Li X J, Tian R Z, Ma C L, Pandey M K, Varshney R K, Wang X J, Zhao C Z. BSA‑seq and genetic mapping identified candidate genes for branching habit in peanut. Theoretical and Applied Genetics, 2022, 135(12): 4457-4468.
[34] Ramirez-Gonzalez R H, Segovia V, Bird N, Fenwick P, Holdgate S, Berry S, Jack P, Caccamo M, Uauy C. RNA-seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnology Journal, 2015, 13(5): 613-624.
[35] Zegeye W A, Zhang Y X, Cao L Y, Cheng S H. Whole genome resequencing from bulked populations as a rapid QTL and gene identification method in rice. International journal of molecular sciences, 2018, 19(12): 4000.
[36] Huang P, Jiang H, Zhu C m, Barry K, Jenkins J, Sandor L, Schmutz J, Box M S, Kellogg E A, Brutnell T P. Sparse panicle1 is required for inflorescence development inand maize. Nature Plants, 2017, 3: 17054.
[37] Zhao Y H, Ma J J, Li M, Deng L, Li G H, Xia H, Zhao S Z, Hou L, Li P C, Ma C L, Yuan M, Ren L, Gu J Z, Guo B Z, Zhao C Z, Wang X J. Whole-genome resequencing-based QTL-seq identifiedgene encoding a R2R3-MYB transcription factor controlling peanut purple testa colour. Plant Biotechnology Journal, 2020, 18(1): 96-105.
[38] Kalendar R, Boronnikova S, Seppänen M. Isolation and purification of DNA from complicated biological samples. Methods in Molecular Biology, 2021, 2222: 57-67.
[39] 邱丽娟,常汝镇. 大豆种质资源描述规范和数据标准. 北京: 中国农业出版社, 2006.
Qiu L J, Chang R Z. Descriptorsand data standard for melon (spp.). Beijing: China Agricuture Press, 2006. (in Chinese)
[40] Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 2009, 25(14): 1754-1760.
[41] McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo M A. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Research, 2010, 20(9): 1297-1303.
[42] Reumers J, De Rijk P, Zhao H, Liekens A, Smeets D, Cleary J, Van Loo P, Van Den Bossche M, Catthoor K, Sabbe B, Despierre E, Vergote I, Hilbush B, Lambrechts D, Del-Favero J. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nature Biotechnology, 2012, 30(1): 61-68.
[43] Cingolani P, Platts A, Wang l L, Coon M, Nguyen T, Wang L, Land S J, Lu X Y, Ruden D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome ofstrain w1118; iso-2; iso-3. Fly, 2012, 6(2): 80-92.
[44] Hill J T, Demarest B L, Bisgrove B W, Gorsi B, Su Y C, Yost H J. MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Research, 2013, 23(4): 687-697.
[45] Fekih R, Takagi H, Tamiru M, Abe A, Natsume S, Yaegashi H, Sharma S, Sharma S, Kanzaki H, Matsumura H, Saitoh H, Mitsuoka C, Utsushi H, Uemura A, Kanzaki E, Kosugi S, Yoshida K, Cano L, Kamoun S, Terauchi R. MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS One, 2013, 8(7): e68529.
[46] 邓泱泱, 荔建琦, 吴松锋, 朱云平, 陈耀文, 贺福初. nr数据库分析及其本地化. 计算机工程, 2006, 32(5): 71-73, 76.
Deng Y Y, Li J Q, Wu S F, Zhu Y P, Chen Y W, He F C. Integrated nr database in protein annotation system and its localization. Computer Engineering, 2006, 32(5): 71-73, 76. (in Chinese)
[47] Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry J M, Davis A P, Dolinski K, Dwight S S, Eppig J T, Harris M A,Hill D P, Issel-Tarver L, Kasarskis A, Lewis S, Matese J C, Richardson J E, Ringwald M, Rubin G M, Sherlock G. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nature Genetics, 2000, 25(1): 25-29.
[48] Tatusov R L, Galperin M Y, Natale D A, Koonin E V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Research, 2000, 28(1): 33-36.
[49] Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Research, 2004, 32 (Supp_1): D277-D280.
[50] Lu N, Rao X L, Li Y, Jun J H, Dixon R A. Dissecting the transcriptional regulation of proanthocyanidin and anthocyanin biosynthesis in soybean (). Plant Biotechnology Jounal, 2021, 19(7): 1429-1442.
[51] Yuan B Q, Yuan C P, Wang Y M, Liu X D, Qi G X, Wang Y N, Dong L C, Zhao H K, Li Y Q, Dong Y S. Identification of genetic loci conferring seed coat color based on a high-density map in soybean. Frontiers in Plant Science, 2022, 13: 968618.
[52] Amarowicz R, Ronald B P. Legumes as a source of natural antioxidants. European Journal of Lipid Science and Technology, 2008, 110(10): 865-878.
[53] Malenčić D, Cvejić J, Miladinović J. Polyphenol content and antioxidant properties of colored soybean seeds from central Europe. Journal of Medicinal Food, 2012, 15(1): 89-95.
[54] Lindner H, Raissig M T, Sailer C, Shimosato-Asano H, Bruggmann R, Grossniklaus U. SNP-Ratio mapping (SRM): identifying lethal alleles and mutations in complex genetic backgrounds by next-generation sequencing. Genetics, 2012, 191(4): 1381-1386.
[55] Zhao M M, Hu B L, Fan Y W, Ding G M, Yang W L, Chen Y, Chen Y H, Xie J K, Zhang F T. Identification, analysis, and confirmation of seed storability-related loci in dongxiang wild rice (griff.). Genes, 2021, 12(11): 1831.
[56] Liu D, Wei X, Sun D, Yang S, Su H, Wang Z, Zhao Y, Li L, Liang J, Yang L, Zhang X, Yuan Y. An SNP mutation of geneconverts petal color from purple to white in radish (L.). Frontiers in Plant Science, 2021, 12: 643579.
[57] Wambugu P, Ndjiondjop M N, Furtado A, Henry R. Sequencing of bulks of segregants allows dissection of genetic control of amylose content in rice. Plant Biotechnology Journal, 2018, 16(1): 100-110.
[58] Lei L, Zheng H L, Bi Y L, Yang L M, Liu H L, Wang J G, Sun J, Zhao H W, Li X W, Li J M, Lai Y C, Zou D T. Identification of a major QTL and candidate gene analysis of salt tolerance at the bud burst stage in rice (L.) using QTL-seq and RNA-seq. Rice, 2020, 13(1): 55.
[59] Holton T A, Cornish E C. Genetics and biochemistry of anthocyanin biosynthesis. The Plant Cell, 1995, 7(7): 1071-1083.
[60] Winkel-Shirley B. Flavonoid biosynthesis. a colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiology, 2001, 126(2): 485-493.
[61] Jaakola L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends in Plant Science, 2013, 18(9): 477-483.
[62] Xu W J, Dubos C, Lepiniec L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends in Plant Science, 2015, 20(3): 176-185.
[63] Li Y Q, Shan X T, Tong L N, Wei C, Lu K Y, Li S Y, Kimani S, Wang S C, Wang L, Gao X. The conserved and particular roles of theregulatorfromhybrida in flower anthocyanin biosynthesis. Plant and Cell Physiology, 2020, 61(7): 1365-1380.
[64] 高瑞芳.与协同调控大豆种皮颜色的机制解析[D]. 长春: 东北师范大学, 2021.
Gao R F. Mechanism analysis ofandsynergistic regulation of soybean seed coat color[D]. Changchun: Northeast Normal University, 2021. (in Chinese)
[65] Zhang Z Y, Shi Y N, Ma Y C, Yang X F, Yin X R, Zhang Y Y, Xiao Y W, Liu W L, Li Y D, Li S J, Liu X F, Grierson D, Allan A C, Jiang G H, Chen K S. The strawberry transcription factor FaRAV1 positively regulates anthocyanin accumulation by activation of FaMYB10 and anthocyanin pathway genes. Plant Biotechnology Journal, 2020, 18(11): 2267-2279.
[66] Xu Z S, Yang Q Q, Feng K, Yu X, Xiong A S. DcMYB113, a root-specific R2R3-MYB, conditions anthocyanin biosynthesis and modification in carrot. Plant Biotechnology Journal, 2020, 18(7): 1585-1597.
[67] Tian J, Peng Z, Zhang J, Song T T, Wan H H, Zhang M L, Yao Y C.regulates coloration via activatingand later structural genes in ever-red leaf crabapple. Plant Biotechnology Journal, 2015, 13(7): 948-961.
[68] Shimada S, Otsuki H, Sakuta M. Transcriptional control of anthocyanin biosynthetic genes in the Caryophyllales. Journal of Experimental Botany, 2007, 58(5): 957-967.
[69] Yan F, Di S, Takahashi R. CACTA-superfamily transposable element is inserted in MYB transcription factor gene of soybean line producing variegated seeds. Genome, 2015, 58(8): 365-374.
[70] Kovinich N, Saleem A, Arnason J T, Miki B. Identification of two anthocyanidin reductase genes and three red-brown soybean accessions with reduced anthocyanidin reductase-1 mRNA, activity, and seed coat proanthocyanidin amounts. Journal of Agricultural and Food Chemistry, 2012, 60(2): 574-584.
Pigment Identification and Gene Mapping in Red Seed Coat of Soybean
CAO Jie1,2, GU YongZhe2, HONG HuiLong2, WU HaiTao2, ZHANG Xia2, SUN JianQiang3, BAO LiGao4, QIU LiJuan1,2
1College of Life Sciences, Jilin Agricultural University, Changchun 130118;2Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081;3College of Agriculture, Northeast Agricultural University, Harbin 150030;4Agriculture and Animal Husbandry Technology Promotion Center of Inner Mongolia Autonomous Region, Hohhot 010018
【Objective】To identify the key genes controlling anthocyanin synthesis and accumulation, to uncover changes in anthocyanin content of the seed coat during seed development, and the primary anthocyanin components responsible for the red seed coat of Taixingaijiaohong (TXAJH); and to lay the groundwork for a thorough understanding of the regulatory mechanism of red seed coat formation.【Method】Using ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-ESI-MS/MS), the anthocyanin composition and concentration of the yellow seed coat of soybean Suinong 14 (SN14) and the red seed coat of soybean TXAJH at various developmental stages were identified. The potential areas of red testa-related genes were first identified using bulked segregant analysis (BSA) on the recombinant inbred lines (RILs) made by crossing SN14 and TXAJH. Based on this discovery, we performed marker linkage analysis to restrict the candidate intervals and predict the candidate genes, and qRT-PCR to confirm the expression of the anticipated candidate genes.【Result】When seed coats from the four developmental phases of SN14 and TXAJH were analyzed, a total of 12 anthocyanins were discovered. Cluster analysis of total anthocyanins revealed substantial changes in the seed coat's anthocyanin composition between TXAJH and SN14 as well as between TXAJH before and after color development. The anthocyanin content of the SN14 seed coat gradually decreased as the seed developed, whereas the TXAJH seed coat's content increased quickly and remained stable. After the development of the seed coat's color, the anthocyanin contents of SN14 and TXAJH showed highly significant differences, and at the mature stage, the TXAJH seed coat's anthocyanin content was more than 200 times that of SN14. The crimson coloring of the TXAJH seed coat was largely due to cyanidin-3-O-glucoside (Cy-3-glu), peonidin-3-O-glucoside (Pn-3-glu), and petunidin-3-O-glucoside (Pt-3-glu). The candidate interval for the red seed coat gene on chromosome 8 was discovered at 8.66 Mb by BSA-seq association analysis. 27 polymorphic markers were used in the marker linkage analysis, which produced 10 haplotypes and reduced the candidate interval to 702 kb. Nonsynonymous variations in 37 genes between the parents were found during this interval, these include the genes for encode the anthocyanin reductase 1 (), the bHLH transcription factor (and), and the MYB transcript factor (). These genes may be involved in regulating the biosynthesis of anthocyanins, and anthocyanin reductase 1 can convert anthocyanins to proanthocyanidins (PA). The results of gene expression analysis revealed that candidate genes and genes related to the anthocyanin biosynthesis pathway had comparable expression patterns in SN14 and TXAJH, and both were expressed at lower levels in SN14 and at higher levels in TXAJH. It was discovered that there was a significant link between the principal constituents of seed coat anthocyanins and the level of candidate gene expression.【Conclusion】The anthocyanin makeup of SN14 and TXAJH's seed coats differed, and Cy-3-glu, Pn-3-glu, and Pt-3-glu may be to blame for the TXAJH's seed coat's red hue. According to predictions,,,, andwill likely be a candidate gene for the red seed coat, in which,, andmay control a number of anthocyanin biosynthesis pathway genes.
soybean; seed coat color; anthocyanin; BSA-seq; gene mapping; transcription factors

10.3864/j.issn.0578-1752.2023.14.002
2023-03-03;
2023-04-23
国家重点研发计划(2021YFD1201600)、中央级公益性科研院所基本科研业务费专项(S2022ZD02)
曹杰,E-mail:cj291@qq.com。通信作者邱丽娟,E-mail:qiulijuan@caas.cn
(责任编辑 李莉)
