无乳链球菌感染卵形鲳鲹脾脏转录组学分析
2023-08-12卞泽昌陈健安王亚丹黄壮杜春民蔡小辉胥鹏关杰耀
卞泽昌,陈健安,王亚丹,黄壮,杜春民,蔡小辉,胥鹏*,关杰耀,
(1.广西大学 动物科学技术学院 亚热带农业生物资源保护与利用国家重点实验室,广西南宁 530004;2.北部湾大学 海洋学院 广西北部湾海洋生物多样性养护重点实验室,广西钦州 535011)
卵形鲳鲹(Trachinotus ovatus)因肉质鲜美、繁殖周期短、经济价值高而在中国被广泛养殖。但近年来,随着卵形鲳鲹养殖规模的不断扩大,病害频繁暴发,链球菌病就是其中之一[1]。链球菌病的频繁发生给世界各地的养鱼业造成了严重的经济损失[2-4],受链球菌影响的鱼类包括牙鲆(Paralichthys olivaceus)[5]、尼罗罗非鱼(Oreochromis niloticus)[6]、斑点叉尾鮰(Ictalurus punctatus)[7]、卵形鲳鲹[3]和观赏鱼[3,8]等,感染链球菌的鱼通常表现出非常相似的症状,如眼球突出、游泳异常、脑膜炎和内脏出血等[2-3]。无乳链球菌作为链球菌中的一种,不仅会影响哺乳动物[9],还会对鱼类造成感染,如齐口裂腹鱼(Schizothorax prenanti)[10]、尼罗罗非鱼(Oreochromis niloticus)[11]和尖吻鲈(Lates calcarifer)[12]等。研究显示无乳链球菌引起的疾病可造成卵形鲳鲹的高死亡率,严重阻碍卵形鲳鲹养殖产业的快速发展[3]。利用分子生物学技术研究卵形鲳鲹感染无乳链球菌后的免疫应答机制是至关重要的。
转录组测序(RNA-seq)作为一种快速、方便的基因组研究方法,可以识别许多功能基因和分子标记[13],近年来已发展成为研究鱼类病毒性和细菌性疾病发病机制的有力工具。例如,利用RNA-seq 技术检测了牙鲆淋巴囊肿病毒、病毒性出血败血症和细胞肿大病毒引起的免疫应答和感染[13-14],以及通过RNA-seq 分析罗非鱼和斑马鱼感染无乳链球菌后的免疫反应[11,15]等。本研究通过给健康的卵形鲳鲹腹腔注射无乳链球菌进行攻毒,然后对卵形鲳鲹脾脏进行RNA 测序。构建测序数据库后进行分析,利用GO 和KEGG 富集分析对差异表达基因(DEGs)和信号通路进行富集分析,并通过STEM 软件分析,确定DEGs 的表达趋势。本研究为进一步筛选和研究卵形鲳鲹无乳链球菌感染后的免疫相关基因和通路提供了理论依据。
1 材料与方法
1.1 实验鱼和菌株
36 条健康的卵形鲳鲹购自广西钦州市犀牛角镇鸿钰水产科技有限公司,体重为(50±0.7)g。并将其置于200 L 水箱中用处理过的海水在28 ~30 ℃下适应10 d。无乳链球菌由北部湾大学蔡小辉副教授惠赠。
1.2 试剂与仪器
TRNzol Universal 总RNA 提取试剂(100 mL),北京天根生化科技有限公司;脑心浸出液肉汤(BHI)(250 g),北京索莱宝科技有限公司;琼脂粉(250 g),上海生工生物工程股份有限公司;无水乙醇、氯仿,分析纯,成都科龙化工试剂有限公司;NaoDrop2000 核酸浓度检测仪,美国Thermo Scientific 公司;DYY-6C凝胶电泳仪,北京六一生物科技有限公司;GDBL-1000 凝胶成像仪,美国Axygen 公司;THZ-92CS 气浴恒温振荡器,杭州川恒实验仪器有限公司;2-16KL台式低温离心机,德国Sigma 公司。
1.3 细菌培养及组织采集
将实验菌株无菌接种到BHI 固体培养基中过夜,然后挑取单克隆至10 mL BHI 液体培养基中,在37 ℃下以200 r/min 振荡孵育过夜,最后在50 mL BHI液体培养基中按1 ∶100(菌液∶液体培养基)体积比例继续在37 ℃下以200 r/min 振荡孵育10 h。4 ℃离心收集细菌菌体,用PBS 缓冲液(pH=7.4)重悬,通过梯度稀释法将重悬菌液稀释106倍,最后使用平板计数法进行计数。
将36 条鱼分为0 h(对照组,C)、6 h(实验组,W6)和24 h(实验组, W24)3 组,每组包含12 条鱼;对照组腹腔注射100 μL PBS 缓冲液(pH = 7.4),W6 组与W24 组腹腔注射100 μL 1×107CFU/mL 无乳链球菌悬液。注射后0 h、6 h 和24 h 收集每组9条鱼的脾脏,每3个脾脏混合成一个样品并标明名称,置于-80 ℃保存至RNA 提取过程。
1.4 RNA 提取、文库构建和测序
使用Trizol 提取总RNA,通过琼脂糖凝胶电泳和核酸浓度检测仪评估RNA 完整性和浓度,之后构建cDNA 文库,送往广州基迪奥生物科技有限公司测序。
1.5 组装和注释
为了保证数据质量,对测序结果进行数据过滤,以减少无效数据所带来的分析干扰,首先利用fastp对raw reads 过滤低质量数据得到clean reads,再通过Trinity 软件进行组装获得unigenes。将获得的unigenes通过Nr数据库、Swiss-Prot蛋白数据库、KEGG数据库、GO 数据库、COG/KOG 数据库进行注释。
1.6 差异表达基因分析方法
使用DESeq2 软件分析3 个不同组之间的差异表达(并在两个样本之间使用edgeR)。以错误发现率(FDR)<0.05 和绝对折叠变化≥2 为参数,判断存在差异表达的基因。基于Wallenius 非中心超几何分布,GOseq 包对差异表达基因进行GO 富集分析。通过KEGG 显著富集分析找出基因显著富集的通路,通过通路显著富集确定基因参与的最主要生化代谢通路和信号转导通路。
1.7 趋势分析
为检验DEGs 的表达模式,先将每个样本的表达数据归一化为0、log2(v1/v0)、log2(v2/v0),再通过STEM 软件进行聚类。随后,对所有图谱中的DEGs 进行GO 和KEGG 富集分析。通过显著性分析和FDR 校正假设检验,将Q 值≤0.05 的GO 条目或通路分类为显著富集的GO 条目或通路。
2 结果与分析
2.1 RNA 测序和功能注释
测序结果显示,raw reads 经过过滤后,所有clean reads 被组装成72 857 个unigenes,其中平均长度和N50 分别为1 121 bp 和2 553 bp。通过主要功能数据库(NR、KOG/COG、Swiss-Prot 和KEGG)共对72 857 个unigenes 进行了注释,获得结果分别为25 089 个(占34.56%)、23 324 个(占32.01%)、18 500 个(占25.39%)和14 712 个(占20.19%)unigenes。通过DESeq2 分析共发现了10 014 个DEGs。相较于对照组,6 h 中发现了4 749 个DEGs,其中上调和下调DEGs 分别为1 812 和2 937 个(图1a);24 h 中共发现8 651 个DEGs,其中上调和下调DEGs 分别为4 418 和4 233 个(图1b)。在6 h 和24 h 组中同时出现表达的DEGs 有3 386 个。

图1 差异表达火山图
2.2 GO 和KEGG 富集分析
基于GO 注释,进行GO 分析以得知DEGs 的功能,GO 分析的3 个主要类别是分子功能(Molecular Function)、细胞成分(Cellular Component)和生物过程(Biological Process)(图2)。在生物过程类别中,6 h 和24 h 的DEGs 主要在细胞过程(Cellular process)、单有机体过程(Single organism process)和代谢过程(Metabolic process)中富集,其中免疫系统过程(Immune system process)在6 h 和24 h 组中同时出现。对于细胞成分类别,两个时间点的DEGs 富集在细胞(Cell)、细胞部分(Cell part)和膜(Membrane)等条目中。在分子功能类别中,两个时间点的DEGs 主要富集在结合(Binding)、催化活性(Catalytic activity)和分子传感器活动(Molecular transducer activity)等条目中。
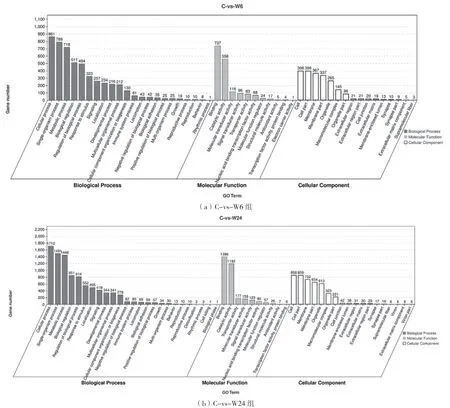
图2 GO 富集分类柱状图
KEGG 富集分析显示,在6 h 和24 h 分别确定了30 个和8 个显著富集的通路(图3)。在6 h 显著富集的前3 条通路主要有细胞因子-细胞因子受体相互作用(Cytokine-cytokine receptor interaction)、疟疾(Malaria)和炎症性肠病(Inflammatory bowel disease)等。在24 h 显著富集的前3 条通路包括蛋白酶体(Proteasome)、溶酶体(Lysosome)和细胞因子-细胞因子受体相互作用(Cytokine-cytokine receptor interaction)。在6 h 和24 h 组中同时发现了细胞因子-细胞因子受体相互作用和类风湿性关节炎(Rheumatoid arthritis)两个通路。
2.3 趋势分析
利用STEM 分析,将20 691 个DEGs 聚类为8 个剖面图,其中0、1、3 和7 这4 个剖面显著聚类(P<0.05),分别包含3 860 个、4 116 个、3 463 个和2 444 个DEGs(图4)。通过KEGG 富集分析从这4 个剖面中发现了7 条主要免疫相关通路,其中产生IgA 的肠道免疫网络、Th1 和Th2 细胞分化、白细胞经内皮迁移、造血细胞谱系、Th17 细胞分化和T 细胞受体信号通路6 条通路显著下调,仅胞质DNA 传感通路显著上调。
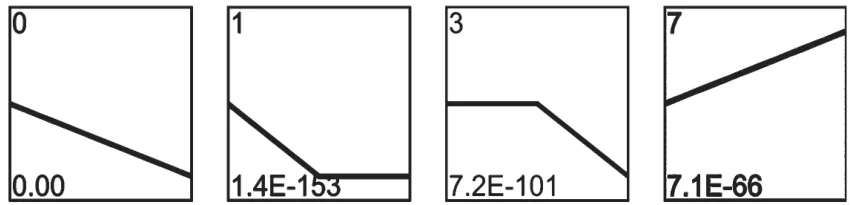
图4 基因趋势图
3 讨论与结论
本研究构建了卵形鲳鲹感染无乳链球菌后的脾脏转录组图谱。研究结果显示,无乳链球菌感染改变了10 014 个基因的表达,在感染6 h 处理组中发现4 749个DEGs,其中上调和下调基因数量分别为1 812个和2 937 个;在感染24 h 处理组中发现8 651 个DEGs,其中上调和下调基因数量分别为4 418 个和4 233 个。通过STEM 分析发现,DEGs 被聚类为8 个趋势图谱,其中0、1、3 和7 四个图谱显著聚集,在这4 张显著富集的图谱中发现了7 条主要与免疫相关的信号通路,其中产生IgA 的肠道免疫网络、Th1 和Th2 细胞分化、白细胞经内皮迁移、造血细胞谱系、Th17 细胞分化、T 细胞受体信号通路6 条通路显著下调,仅胞质DNA 传感通路显著上调。在罗非鱼无乳链球菌感染的研究中,早期时间点的DEGs 被快速诱导,晚期时间点的DEGs 表达水平逐渐下降,表明罗非鱼可能具有快速识别入侵细菌并通过激活几种免疫相关途径来清除细菌的强大能力[11]。与罗非鱼研究结果不同,本研究中大多数基因在早期迅速下调,在后期逐渐稳定,这可能意味着卵形鲳鲹在早期缺乏快速识别和抵抗细菌入侵的能力,导致细菌在早期抑制了大多数免疫相关基因和通路,使其无法发挥免疫作用。本研究结果为进一步筛选卵形鲳鲹无乳链球菌病的免疫关键基因和通路提供了基础信息,为卵形鲳鲹疾病预防和治疗提供了新见解。
