仅脑脊液抗体阳性的髓鞘少突胶质细胞糖蛋白抗体相关疾病1例报告及文献复习
2023-08-10裴晓晨秦毅丹陈加俊
裴晓晨, 李 佳, 秦毅丹, 苏 杭, 陈加俊
髓鞘少突胶质细胞糖蛋白抗体相关疾病(MOG antibody disease,MOG-AD)是中枢神经系统的一种炎症性脱髓鞘疾病,临床特征为单相或复发相的神经功能障碍,此类疾病不符合多发性硬化及其他已知神经炎症疾病的典型标准。多数MOG-AD患者血清中MOG抗体呈阳性、脑脊液中为阴性。目前仅有少数报道该疾病患者MOG抗体仅在脑脊液中呈阳性。然而,我们在临床中发现1例少见的MOG-AD患者,该患者脑脊液中MOG抗体为弱阳性(1∶10),血清中抗体为阴性,结合影像及其他相关辅助检查,考虑该患者免疫反应始发于颅内的胶质细胞等部位,由硬脑膜窦中的抗原提呈细胞(APC)捕获脑脊液中的MOG抗原后产生一系列免疫反应,从而产生MOG抗体[1]。仅在脑脊液中MOG抗体阳性的患者与外周血中MOG阳性、脑脊液中MOG阳性/阴性的MOG-AD患者相比,可能血脑屏障未遭受破坏,提示免疫反应可能仅发生在颅内,具体机制目前尚未明确。该患者接受糖皮质激素冲击治疗后,逐渐递减激素剂量直至停药,患者脑脊液MOG抗体转阴,病灶明显减小,患者现恢复良好,未出现复发。
1 病例资料
患者,女,51岁,入院1年前在无明显诱因的情况下开始出现饮水呛咳及小便失禁,入院15 d前自觉右侧肢体麻木,表现为“蚁走感”;12 d前出现右下肢无力,逐渐加重,无法独立行走,而后相继出现右上肢无力,不能持物;3 d前自觉气短,说话时需深呼吸,患者上述症状逐渐加重,来吉林大学中日联谊医院诊治。患者病程中伴有言语不清、饮水返呛、左侧头痛、大便费力、有时小便失禁。该患者否认相关遗传疾病的家族病史。
1.1 入院时神经系统查体 意识清醒,构音障碍,左侧咽反射迟钝,右侧肢体肌力3级,左侧肢体肌力5级,四肢腱反射活跃,四肢肌容积正常,双侧指鼻试验不稳准,双侧快速轮替试验笨拙、双侧跟膝胫试验不稳准,右侧肢体痛温觉、位置觉减退,双侧Babinski、Chaddock征阳性,余神经系统查体未见异常。
1.2 影像学检查结果 患者磁共振成像(MRI)检查结果显示,该患者病灶表现为延髓内边界模糊的斑片状异常信号影,中脑也存在可疑改变,病变性质考虑为脱髓鞘病变可能性大,待除外占位。(A)~(G)为入院时MR结果,病灶面积较大、信号较高;(H)~(I)为治疗12 d后复查MR结果,病灶面积缩小、信号减低(见图1)。

图A、B 头部MR平扫+弥散提示:延髓见斑片状异常信号影,T1WI呈等、低信号,T2WI及FLAIRA呈高信号,DWI未见受限,T2WI矢状位似见中脑少许稍高信号影;图C~E 磁共振头部普通增强提示:延髓见结节状、斑片状异常信号影,呈不规则环形强化,大小约(0.5~1.8)×1.0 cm;图F、G 颈椎MR平扫提示:延髓内见斑片状稍长T1,稍长T2异常信号,压脂像呈稍高信号。颈4-5间盘水平硬膜囊后缘见低信号压迹;图H 颈椎MR平扫提示:延髓内见斑片状稍长T1,稍长T2异常信号,压脂像呈稍高信号;图I 头部MR平扫提示:延髓内可见不规则混杂长T1混杂长T2信号影,FLAIR呈高信号。

1.3 入院后病情变化 患者入院后第2天呼吸困难加重,主要表现为气短、呼吸无力。患者在低流量吸氧状态下血氧饱和度维持在98%,但咳嗽力量尚可,血气分析:SO298%。结合患者病史、症状特点及目前辅助检查,考虑患者延髓及高颈段病变为脱髓鞘可能性大,但不能除外为延髓肿瘤性病变(见图1A~G)。神经外科会诊后考虑患者病变位置特殊,无法进行活检及手术治疗。因此与患者及家属沟通后,考虑暂时按照脱髓鞘病变进行治疗,给予患者甲泼尼龙静脉滴注治疗(1000 mg·d-1,持续3 d,每3 d减半)。与此同时,进行视神经诱发电位检查:VEP:两眼分别刺激,双侧各波波形分化可,双侧N75、P100、N145潜伏期正常,波幅正常。提示:双侧视觉径路传导未见异常,患者视力一直未受影响。并完善腰椎穿刺术,脑脊液为无色清亮液体,测得脑脊液压力90 mmH2O,脑脊液常规、生化提示脑脊液白细胞计数 14×106/L。血常规、凝血四项、肝功肾功、贫血两项、同型半胱氨酸未见明显异常。随后化验血清及脑脊液多发性硬化、视神经脊髓炎等相关抗体指标。结果提示,脑脊液中MOG抗体为弱阳性(1∶10),血清中抗体均为阴性(见图2、图3)。患者使用甲泼尼龙治疗后病情逐渐缓解,2 d后呼吸困难、右侧肢体麻木、无力逐渐有所好转。

脑脊液的起始稀释梯度为1∶1,血清的起始稀释梯度为1∶10,加号数量代表该稀释度下的荧光强度;通过基于细胞转染的间接免疫荧光法(cell based assay,CBA)进行检测。
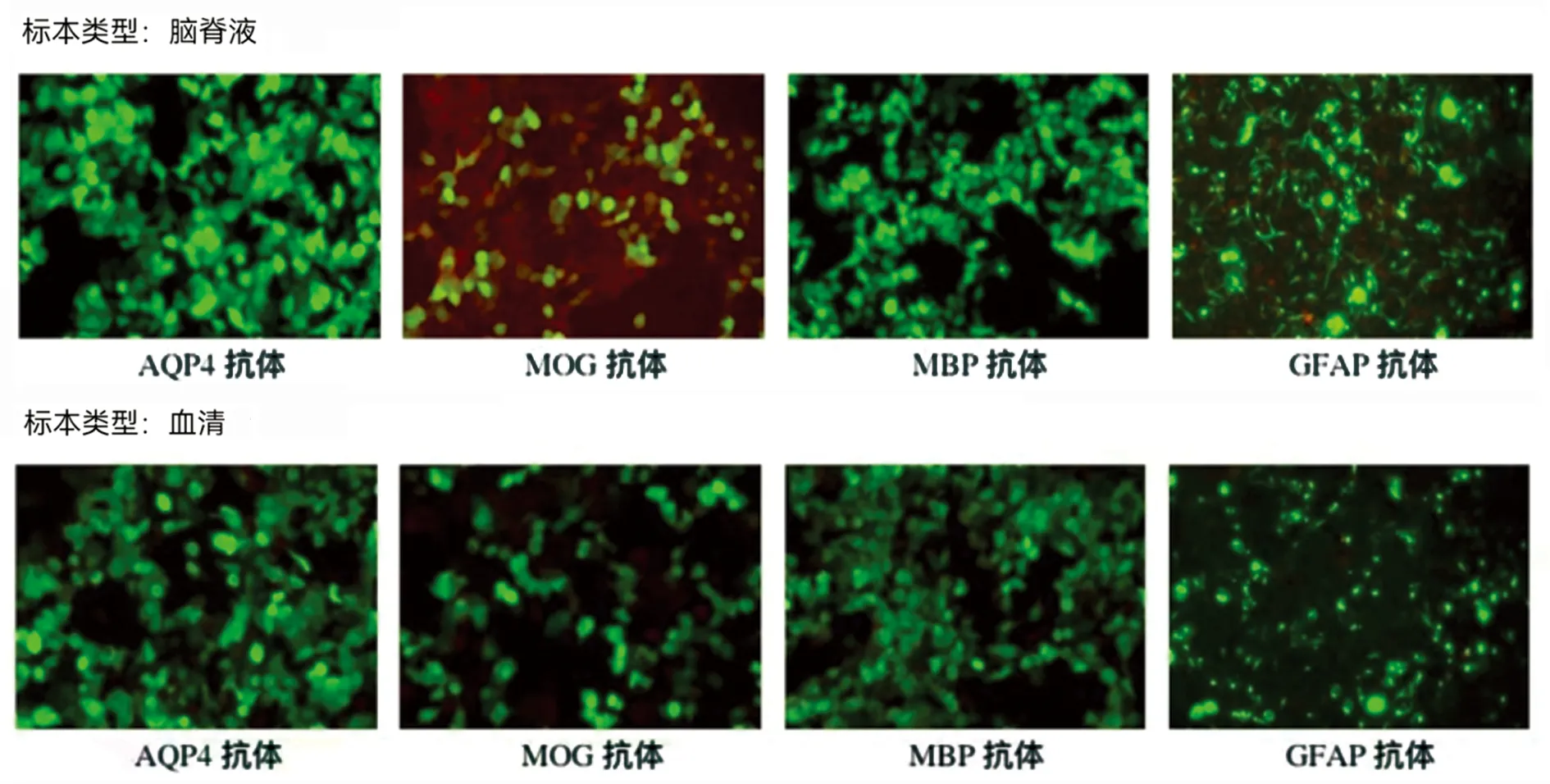
图3 神经免疫检测荧光图片
使用糖皮质激素治疗后12 d,复查脑脊液、血清相关抗体均为阴性,脑脊液常规生化提示脑脊液糖 4.1 mmol/L。复查头部MR提示病灶范围略有缩小(见图1H、图1I)。且患者右侧上下肢肌力有明显改善,出院时肌力4级+。出院后继续规律口服泼尼松龙,无言语不清、肢体麻木及无力。出院4个月,患者复查头部MR提示病灶范围较前缩小,患者恢复良好,暂未出现复发。
2 讨 论
髓鞘少突胶质细胞糖蛋白(MOG)是少突胶质细胞产生的位于髓鞘表面上的蛋白质之一,少突胶质细胞是中枢神经系统中的形成髓鞘的细胞,MOG是少突胶质细胞膜表面的重要组成部分;在髓鞘的形成,维持和分解中具有基本作用,且MOG只在中枢神经系统被发现,可以起到细胞黏附分子的作用,参与调节少突胶质细胞微管稳定性并介导补体级联反应,通过MOG的黏附功能和髓鞘与免疫系统之间相互作用的维持髓鞘的结构完整性[2,3]。
近年来,已经证实,一些血清水通道蛋白4(AQP4-IgG)抗体阴性的视神经脊髓炎谱系疾病(NMOSD)患者具有抗髓鞘少突胶质糖蛋白(MOG)的IgG抗体,被诊断为MOG-AD[MOG-IgG的相关性脑脊髓炎(MOG-EM)或MOG-IgG的自身免疫病],是一种中枢神经系统(CNS)炎性脱髓鞘疾病,不同于多发性硬化症(MS)和血清AQP4-IgG阳性神经脊髓炎频谱障碍(AQP4-IgG-NMOSD),不符合MS或其他已知神经炎性疾病的典型标准,最常见的是视神经炎(ON),其次是脊髓炎,急性播散性脑脊髓炎(ADEM)或ADEM样呈现(例如,脑干攻击),其特征是神经功能障碍的单相或复发性过程[2,4,5]。
据以往研究,MOG抗原可能是在血-脑屏障的通透性增加时(如CNS感染时)渗漏入外周而被外周免疫系统识别,通过肠道菌群辅助MOG特异性地CD4+T细胞和B细胞生成和活化,促进了MOG抗体在外周淋巴器官中产生。MOG抗体为炎性脱髓鞘疾病的致病性抗体,血脑屏障的功能再次遭受破坏后,MOG-IgG通过被动扩散或通过渗漏的血脑屏障到达CSF中[6]。MOG-Ab与T细胞协同作用后具有同致病能力,被动扩散到达CSF后通过细胞毒作用,诱导补体和抗体介导神经系统损伤[7]。血清MOG-Ab的检测被视为确诊MOG-AD的金标准,然而当血清抗体阴性、临床高度怀疑为NMOSD、MOG、AQP4-Ab病例时,行脑脊液MOG-Ab检测可以为诊断提供帮助。临床上仍有极少数仅在脑脊液中存在MOG-Ab的MOG-AD患者,这类患者的致病抗体诱导产生的B细胞可以仅存在于中枢神经系统中,反映了MOG-Ab在鞘内合成的可能而非血清中的抗体被动扩散至中枢神经系统中[8]。
神经免疫的相互作用可导致许多神经系统疾病的发生,虽然中枢神经系统免疫监视的机制尚不明确,但可能是导致该病发生的重要因素。硬脑膜中存在稳定的白细胞群体,对参与适应性免疫功能的免疫群体进行整体免疫组化研究后发现,T细胞和组织相容性复合体Ⅱ(MHCⅡ)+抗原呈递细胞(APCs)并非均匀分布在整个硬脑膜组织中,而是高度分布在硬脑膜窦周围(鼻窦),成为免疫枢纽。动物实验证明,在使用小剂量示踪剂注射于小鼠纹状体实质内后,最终发现许多维持大脑稳态的蛋白质积累于硬脑膜周围。硬脑膜脉管系统可以进行平稳持续的T细胞监视,硬脑膜窦相关的APCs可以捕获脑脊液抗原,并且硬脑膜窦是循环的自我反应性T细胞遇到其同源抗原的关键部位,包括EAE/MS中的MOG反应性T细胞。在研究自身免疫性脑脊髓炎(EAE)中 CNS 抗原的局部呈递与自身免疫性神经炎症的相关性时,在EAE发病后,MHCⅡ+APC 数量没有显著变化,但在硬脑膜内积累了大量的T细胞,通过液相色谱-质谱(liquid chromatography-mass spectrometry,LC-MS)分析显示硬脑膜存在 MOG 积累。同时,在实验中也观察到了在EAE发病期间MOG 反应性T细胞在外周的激活、在硬脑膜中的积累。在EAE急性发作时,所有CNS脉管系统都被炎性浸润,但T细胞可以从不同部位浸润CNS组织,从而导致不同部位的神经系统疾病[4]。以上机制为MOG-AD存在原发于中枢神经系统的情况提供了有力依据。
放射学和血清学确诊的MOGAD的病理学特征是静脉和融合性白质脱髓鞘。这表明MOG-AD脱髓鞘的方式介于MS和具有静脉型的ADEM之间。炎性浸润主要由CD4+T细胞和粒细胞组成,而CD8+T细胞占主导地位。皮质内的脱髓鞘病变更为常见。在活动性病变中可以观察到补体沉积,但在星形胶质细胞或神经胶质母细胞上未观察到补体沉积,因为MOGAD不是星形细胞病变,使得AQP4未受到损伤,由此可以很容易地与AQP4阳性的NMOSD和MS区分开[3]。对1例仅在CSF中具有MOG-Abs的患者的大脑,脊髓、神经根和软脑膜的尸检后发现,该患者存在大量的组织破坏和脑桥和脊髓的脱髓鞘,其主要特征是相对稀疏的轴突、静脉、皮下及皮质汇合的原发性脱髓鞘,反应性神经胶质增生和主要由粒细胞,巨噬细胞和CD4+T细胞构成的炎性浸润,并在炎性浸润物中,发现了IgG沉积物,证实了仅在CSF中具有MOG-Abs的炎性浸润相关病变确实存在[7]。
虽然该患者中枢神经系统中MOG-Ab的滴度较低,但仍考虑致病性抗原滴度较低也可导致脊髓炎的发生[8~10]。一项对100例髓鞘少突胶质细胞糖蛋白(MOG)抗体患者的脑脊液163次腰椎穿刺的结果研究发现:在少数有鞘内IgG合成的样本中,仅在急性疾病发作期间发现鞘内MOG-IgG,3/4随访的患者CFS中MOG-IgG仅短暂存在。3/6 寡克隆带(OCB)阳性患者中的OCB只是短暂出现一次。此研究说明了对多数MOG-IgG阳性EM患者的CSF进行定性和定量研究后,提示如果鞘内IgG合成存在,合成率往往较低,且主要在急性发作时短暂存在[11]。
据报道,在急性期静脉注射甲泼尼龙(IVMP)可有效治疗MOG抗体阳性ON和(或)脊髓炎(剂量为每天1~2 g,连续3~5 d),患者症状可能得到缓解[2],较严重或类固醇治疗无效的处于疾病急性期的患者应早期进行血浆置换和静脉内免疫球蛋白(IVIG)治疗[3]。该患者急性期经激素冲击疗法治疗12 d后,复查CSF中MOG-Ab由弱阳性转为阴性,从患者症状以及辅助检查都可以体现出急性期甲泼尼龙冲击疗法治疗有效。
对于长期复发的预防性治疗,泼尼松龙的长期治疗(体重>40 kg的患者,剂量>10 mg/d,体重≤40 kg的患者,剂量>5 mg),静脉内免疫球蛋白(诱导疗程剂量为2 g/kg,随后为每月) 1 g/kg输注,利妥昔单抗、霉酚酸酯、氨甲蝶呤或硫唑嘌呤都有报道,以减少MOG-AD复发。但是,利妥昔单抗治疗可能与首次输注后几周内的新发作有关,可能是由于B细胞活化因子和自身抗体水平的暂时升高,正如一些AQP4阳性NMO患者所观察到的。那他珠单抗治疗不能有效地预防复发,而奥法木单抗(ofatumumab)的治疗却能将年复发率从2.1降低至0.66。一项研究发现,与非类固醇维持疗法(38%)相比,接受类固醇维持疗法的患者治疗失败率更低。口服类固醇作为免疫抑制剂辅助治疗的作用尚不清楚。一项研究发现,接受硫唑嘌呤治疗但未辅以口服类固醇治疗的MOG抗体ON和(或)脊髓炎患者比接受联合治疗的患者更容易复发。此外,复发似乎更频繁地发生时口服泼尼松龙的剂量下降到低于10 mg/d或停止类固醇。干扰素β治疗已被证明是无效的,并且可增加患病的风险。同样,醋酸格拉替雷在儿童患者中也被证明无效[2,3]。因此,该患者静脉滴注12 d甲泼尼龙治疗后,改为口服甲泼尼龙片,起始量为60 mg/d,每15 d减4 mg至停药,患者现未出现复发。但尚未发现高度特异性的放射学表现来区分MOGAD和非MOG抗体病例。
3 结 论
该患者仅脑脊液中MOG抗体为阳性,考虑神经免疫反应可能于中枢神经系统的硬脑膜窦中完成,相关抗体未渗入外周。因此,临床上考虑为中枢神经系统炎性脱髓鞘疾病的患者,血清中相关抗体均为阴性时,要注意完善脑脊液中相关抗体检测,以避免漏诊及误诊。
