过量表达气囊蛋白强化茂原链霉菌合成胰蛋白酶
2023-07-27苏莹莹刘松
苏莹莹,刘松*
1(江南大学,未来食品科学中心,江苏 无锡,214122) 2(江南大学,粮食发酵工艺与技术国家工程实验室,江苏 无锡,214122)
气体囊泡是具有刚性空心结构并充满气体的细胞器[1],广泛存在于蓝藻、异养细菌、厌氧光合细菌和嗜盐古菌等原核生物中[2]。一般认为,气体囊泡可为蓝藻细胞提供浮力,从而更好地利用光和养分[3]。在嗜盐古菌中,气体囊泡则有助于其在极端压力和高盐条件下存活[2,4]。结构分析显示,气体囊泡主要由气囊蛋白(gas vesicle protein, Gvp)构成[5],在不同生物体中已经鉴定出8~14个编码可调控气体囊泡形成的基因。嗜盐古菌中,气囊由2个相反的基因簇gvpANO和gvpFGHIJKLM组成[6],GvpA作为大多菌体气囊的主要结构蛋白,高度保守且具有很强的疏水性[7];gvpO则可能参与了gvpACN的转录激活或有助于其mRNA的稳定性,敲除gvpO会显著降低gvpA、gvpC和gvpN的转录水平且无法形成气体囊泡[8]。研究表明,Gvp的表达受环境因素的调节[2]。在沙雷氏肠杆菌(Serratiasp.ATCC 39006)中观察到的气体囊泡,其形成就需要氧气的刺激,并受到群体感应信号分子N-酰基高丝氨酸内酯的调控[9]。值得注意的是,重组表达编码蓝藻气体囊泡基因簇能使大肠杆菌细胞浮于培养基表面[10]。因此,过量表达气体囊泡基因簇能有效调控细胞的生理状态。
在链霉菌基因组中研究者同样发现了gvp基因簇,如天蓝色链霉菌(Streptomycescoelicolor)的gvpOAFGYZJLSK[2-3]。不同于蓝藻和嗜盐古菌,链霉菌gvp基因簇一般缺少gvpC、gvpN和gvpV等气囊蛋白基因。值得注意的是,蓝藻和嗜盐古菌中的辅助蛋白GvpC能使气体囊泡更稳定抗压,GvpN或GvpV则有助于促进气囊组装[9]。因此,多数链霉菌并不能检测到气体囊泡的形成[11],过量表达链霉菌gvp基因簇也不能使大肠杆菌细胞漂浮[12]。一般认为,链霉菌主要是利用GvpA的疏水性N端形成疏水表面,以便于其气生菌丝穿过气液界面[3]。最近,链霉菌(Streptomycessp.) CB03234-S中发现的基因簇gvpOAFGJLSK(图1)缺少了链霉菌中常见的gvpY和gvpZ,并能在富含氮源的培养条件下观测到胞内的类似气囊蛋白的结构[13]。此外,过量表达gvpOAFGJLSK增加CB03234-S中的气体囊泡数量和十元环烯二炔天赐霉素的产量,使其链霉菌菌体形态和代谢产生重要的影响[13]。因此,调控gvpOAFGJLSK的合成可能是促进链霉菌产物合成的新途径。
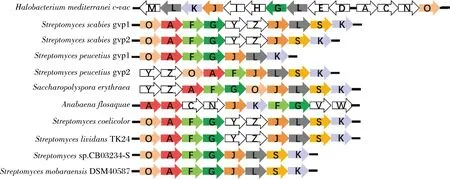
图1 不同来源的gvp基因簇组成Fig.1 Composition of gvp gene clusters from different sources
基因组分析显示,S.mobaraensisDSM40587的gvp基因gvpOAFGJLSK(gvp40587)与CB03234-S的gvp基因簇具有相同的组成(图1),均不含gvpY和gvpZ。在本研究中,将S.mobaraensisDSM40587胰蛋白酶(S.mobaraensistrypsin,SMT)过量表达于缺失谷氨酰胺转氨酶基因(tg)的S.mobaraensisDSM40587(smY2019Δtg)中[14],研究了过量表达gvpA、gvpO及基因簇gvp40587对SMT合成、生长及代谢的影响。研究结果将有助于认识Gvp功能,也为链霉菌产酶的优化提供参考。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
S.mobaraensisDSM40587为本研究室保藏。tg缺失的S.mobaraensis(smY2019Δtg)为研究室前期工作中构建[14]。大肠杆菌(EscherichiacoliJM109)为本章实验所用克隆宿主。本研究使用的其他菌株与质粒如表1所示。
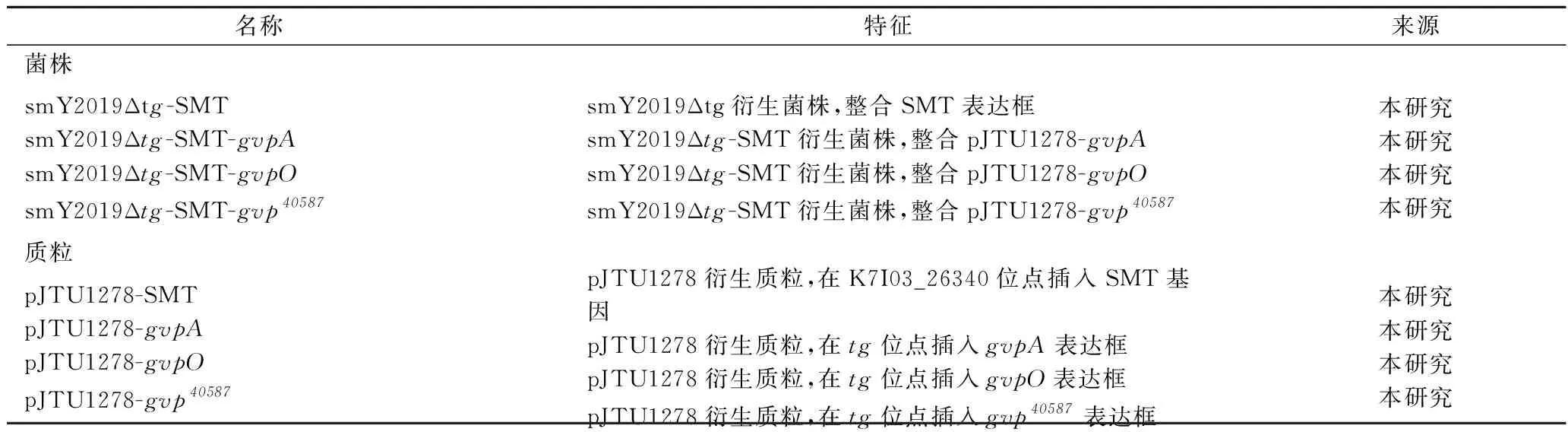
表1 本研究所使用的菌株与质粒Table 1 Strains and plasmids used in this study
1.1.2 试剂
高保真DNA聚合酶(PrimeSTAR GXL)、平末端磷酸化连接酶(Blunting Kination Ligation Kit),宝生物工程(大连)有限公司;高保真DNA聚合酶(KOD FX),东洋纺(上海)生物科技有限公司;一步克隆连接试剂盒One Step Cloning Kit(ClonExpressTMII),南京诺唯赞生物科技股份有限公司;限制性内切酶、蛋白预制胶、蛋白质Marker、MES蛋白电泳缓冲液,赛默飞世尔科技(中国)有限公司;硫链丝菌素,生工生物工程(上海)股份有限公司;质粒抽提试剂盒、DNA产物纯化试剂盒、基因组提取试剂盒、ATP含量检测试剂盒,生工生物工程(上海)股份有限公司;NADPH/NADP+测定试剂盒,碧云天生物技术有限公司;底物Nα-苯甲酰-DL-精氨酸-p-硝基苯酰胺(BAPNA),美国Sigma公司;RNA提取试剂盒,天根生化科技(北京)有限公司;反转录试剂盒和荧光定量PCR试剂盒,宝生物工程(大连)有限公司。其他常规试剂及药品为国产或进口分装。
1.1.3 培养基
LB培养基(g/L):胰蛋白胨10,酵母提取物5,NaCl 10。
GYM产孢培养基(g/L):葡萄糖10,酵母粉4,麦芽粉3。
2×YT孢子萌发培养基(g/L):蛋白胨16,酵母粉10,NaCl 5。
种子培养基(g/L):甘油20,酵母粉5,胰蛋白胨20,K2HPO44,MgSO42,pH 7.2。
发酵培养基(g/L):甘油20,酵母粉5,胰蛋白胨20,大豆粉20,KH2PO42,K2HPO44,MgSO42,CaCO32,pH 7.2~7.4。
固体培养基在此基础上添加1.5%~2%琼脂粉。121 ℃湿热灭菌15 min后备用。
1.2 实验方法
1.2.1 整合表达载体的构建
以S.mobaraensisDSM40587基因组(GenBank No.CP083590)为模板,分别扩增基因K7I03_26340上游1 056 bp(同源重组左臂)和下游999 bp(同源重组右臂)、SMT基因K7I03_25045片段(792 bp)和tg启动子片段。使用限制性内切酶BamH I和EcoR I切割pJTU1278获得同源重组质粒片段。采用一步克隆试剂盒将上述5个片段融合,得到SMT整合表达质粒pJTU1278-SMT。以S.mobaraensisDSM40587基因组为模板,分别扩增tg位点上游1 406 bp(同源臂)、相关气囊基因gvpA、gvpO及完整基因簇gvp40587。采用一步克隆试剂盒将gvpA、gvpO及完整基因簇gvp40587分别与tg同源臂和pJTU1278酶切片段连接,得到整合表达质粒pJTU1278-gvpA、pJTU1278-gvpO和pJTU1278-gvp40587。将重组质粒转化E.coliJM109,并进行PCR验证。载体构建及验证相关引物见表2。

表2 本研究所使用的引物Table 2 Primers used in this study
1.2.2S.mobaraensis原生质电转化
取70 μLS.mobaraensis原生质体溶液与5~10 μg构建的整合表达质粒混合,转入0.1 cm电转杯中,于2 500 V和25 Ω电击1次;电击结束立即加入1 mL预冷的1 mol/L山梨醇,最后转移至无菌的1.5 mL EP管中,于30 ℃、220 r/min摇床中培养2 h。将培养液涂布至含30 μg/mL硫链丝菌素的GYM平板,培养5~6 d筛选含单交换的转化子。将转化子转接至无抗性GYM平板松弛培养3~4 d后,通过PCR验证和基因测序分析,筛选获得双交换转化子。相关PCR验证引物见表2。
1.2.3 重组S.mobaraensis摇瓶发酵
将GYM平板上获得的重组菌转化子平板上划线后,置于30 ℃恒温培养箱内培养4~5 d。随后用无菌涂布棒适量刮取GYM固体平板上的菌丝,将孢子悬液以终浓度106个/mL接种至种子培养基(30 μg/mL硫链丝菌肽),于30 ℃和220 r/min摇床中培养24~36 h后,以8%(体积分数)接种量转接至含有30 mL发酵培养基(30 μg/mL硫链丝菌肽)的250 mL三角瓶中,继续于30 ℃和220 r/min摇床中培养72 h。
1.2.4 胰蛋白酶酰胺酶活力测定
每隔12 h,将不同时间点的发酵样品取1 mL于10 000 r/min、10 min进行离心,之后取上清至EP管中,放冰上待测。参考文献[15]的方法进行测定:以BAPNA为底物测定胰蛋白酶酰胺酶活力:将890 μL缓冲液溶液(50 mmol/L pH 8.0 Tris-HCl, 20 mmol/L CaCl2)与10 μL BAPNA (100 mmol/L)混合均匀;在37 ℃下预热10 min后,迅速加入100 μL酶液混合均匀,在37 ℃、410 nm条件下测定其吸光值的变化值(ΔA410/min)。酶活定义为:在37 ℃下,ΔA410/min升高0.1即为胰蛋白酶的1个酰胺酶水解单位。计算如公式(1)所示:
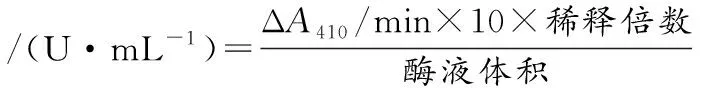
(1)
1.2.5 SDS-PAGE凝胶电泳分析
取30 μL发酵上清液与10 μL的5xloading buffer混合均匀后,于90 ℃加热10 min后待用。SDS-PAGE凝胶电泳具体操作方法参考Invitrogen公司Bis-Tris预制凝胶说明书。
1.2.6 实时荧光定量PCR(qRT-PCR)
取基础发酵培养基中培养36 h的发酵液,于4 ℃、12 000 r/min、2 min离心,收集菌体待用,随后用RNA提取盒提取总RNA,根据说明书中的步骤进行操作。对提取RNA的浓度、纯度及完整性进行验证,并以RNA为模板,使用PrimeScriptTMRT试剂盒逆转录成cDNA,最终,在Light Cycler 480 System(Roche Diagnostics Ltd.,Rotkreuz,Switzerland)中使用SYBR Green I嵌合荧光法对qRT-PCR进行分析。以S.mobaraensis基因组中的hrdB基因为内参基因,用2-ΔΔCt法计算不同样品中不同基因的相对倍数变化。qRT-PCR使用的引物列于表3中。
对于局部损坏的素土后屋面,采用干土+草泥方式修补。对于完全需要重新改造的素土后屋面,拆除修平后,在原屋面上铺设聚苯板保温层+塑料薄膜或草泥抹面的简易厚屋面。有立柱温室和骨架荷载良好的温室,采用垫板+聚苯板+钢网砼层,垫板+聚苯板+CSM墙面材料等复合后屋面。对于原屋面为水泥抹面面层时,如裂缝、剥落现象,应将原墙面凿毛,否则,应将原涂层铲除,重新进行水泥抹面,裂缝可采用防水沥青灌缝处理。后屋面改造升级后,达到整个后屋面顶部成南高北低的斜坡,坡面平整无缝,具备防水、隔热保温的功能,并在后屋面加盖保温被,或覆盖棚膜等方式,进行防水保温。

表3 qRT-PCR所用引物Table 3 Primers used for qRT-PCR
1.2.7 菌丝生长量测定
将不同时间点的发酵样品取1 mL进行离心,留下菌体,每隔12 h取样1 mL至1.5 mL提前称重的空EP管(m1)中。将菌液离心,10 000 r/min、10 min,留下菌体,EP管开盖放至65 ℃烘箱中干燥直到恒重,取出称量(m2)。计算菌体在不同时间点的质量(m2-m1),绘制细胞干重曲线。
1.2.8 ATP测定
将不同时间点的发酵样品取1 mL进行离心,留下菌体,用PBS缓冲液(137 mmol/L NaCl,2.7 mmol/L KCl,10 mmol/L Na2HPO4,1.8 mmol/L KH2PO4,pH 7.4)洗涤3次后进行超声破碎1 min,随后10 000 r/min、4 ℃离心10 min,取上清至另一EP管中,加入500 μL氯仿充分振荡混匀,10 000 r/min、4 ℃离心3 min,取上清至冰上待测,胞内ATP测定按ATP检测试剂盒说明书操作。
1.2.9 NADPH/NADP+测定
将不同时间点发酵样品取1 mL进行离心,留下菌体,用PBS洗涤3次后加入适量预冷的NADP+/NADPH提取液,轻轻吹打促进细胞裂解,随后10 000 r/min、4 ℃离心10 min,取上清至冰上待测,NADPH/NADP+测定按辅酶II NADP(H)含量试剂盒说明书操作。
1.2.10 透射电镜分析
2 结果与分析
2.1 胰蛋白酶在S.mobaraensis中的整合表达
胰蛋白酶是一种重要的碱性丝氨酸蛋白酶[15],广泛应用于皮革、纺织、制药和食品等行业[16]。为实现胰蛋白酶的高效表达,将S.mobaraensisDSM40587来源的胰蛋白酶SMT完整的开放阅读框与tg启动子和K7103_26340位点的左右同源臂融合并克隆至pJTU1278,构建得到SMT整合表达质粒pJTU1278-SMT(图2-a)。将pJTU1278-SMT转化smY2019Δtg,通过PCR验证和基因测序筛选得到在K7103_26340位点整合SMT表达框的smY2019Δtg-SMT(图2-a、图2-b)。
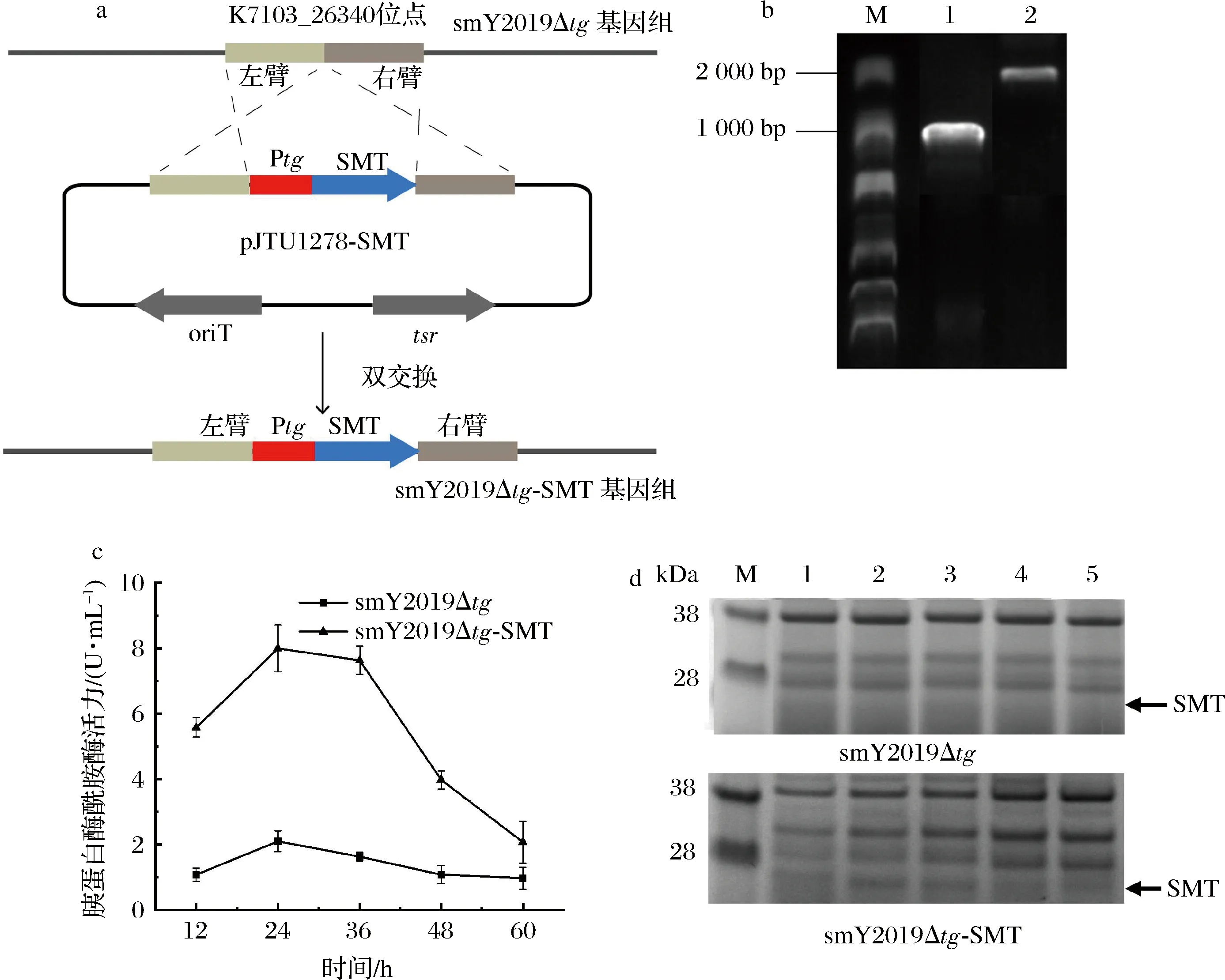
a-双交换整合胰蛋白酶基因示意图;b-DNA核酸电泳图(M-DNA标准分子质量;1-smY2019Δtg;2-smY2019Δtg-SMT);c-重组菌发酵过程酶活力分析;d-重组菌发酵过程蛋白电泳分析(M-蛋白marker;1-12 h;2-24 h;3-36 h;4-48 h;5-60 h)图2 胰蛋白酶在smY2019Δtg中的表达分析Fig.2 The analysis of trypsin expression in smY2019Δtg
分别对smY2019Δtg-SMT和smY2019Δtg发酵,测定胰蛋白酶酰胺酶活力和进行SDS-PAGE电泳分析。酶活力分析显示,在发酵上清液中胰蛋白酶活力在24 h达到最高值8.0 U/mL,发酵36 h后活力迅速下降(图2-c)。尽管未过量表达SMT,smY2019Δtg发酵上清液仍能检测到胰蛋白酶活力,并在发酵24 h达到最大值2.2 U/mL。SDS-PAGE分析显示,smY2019Δtg-SMT的24 h和36 h发酵上清液有较明显的SMT条带(理论分子质量26 kDa)(图2-d),而在smY2019Δtg发酵上清液中未见明显的SMT条带。上述结果表明,smY2019Δtg内源的SMT具有一定的表达量,采用tg启动子在K7103_26340位点整合表达SMT能显著提升其胞外表达水平。
2.2 过量表达gvp基因对胰蛋白酶发酵的影响
目前,Gvp在胞内的组装机制尚不清楚,相关基因在不同宿主中的功能存在一定的差异[17]。例如,过量表达结构蛋白GvpA和辅助蛋白GvpC不能促进嗜盐古菌中气体囊泡形成[18],而表达蓝藻Anabaenaflos-aquae和Planktothrixrubescens中GvpA和GvpC则能使酿酒酵母中产生气体囊泡[19]。为分析气体囊泡对SMT分泌表达的影响,通过单交换在smY2019Δtg-SMT中tg位点整合表达内源gvpA、gvpO和完整基因簇gvp40587,分别得到重组菌smY2019Δtg-SMT-gvpA,smY2019Δtg-SMT-gvpO和smY2019Δtg-SMT-gvp40587(图3-a)。
将构建的重组菌进行发酵,胰蛋白酶酰胺酶活力和SDS-PAGE电泳分析SMT表达情况。结果显示,smY2019Δtg-SMT-gvp40587发酵到36 h胞外胰蛋白酶活达到最大值13.14 U/mL,较smY2019Δtg-SMT最高酶活(24 h)提高了64.3%;整个发酵过程的smY2019Δtg-SMT-gvp40587胞外酶活力均明显高于smY2019Δtg-SMT,后期酶活下降速度相对较慢(图3-b)。与smY2019Δtg-SMT相同,smY2019Δtg-SMT-gvpA胞外最高胰蛋白酶活在24 h达到最高值,但较前者提高17%(图3-b)。如图3-c所示,smY2019Δtg-SMT-gvp40587和smY2019Δtg-SMT-gvpA发酵上清液胰蛋白酶条带较smY2019Δtg-SMT明显增粗(图3-d)。然而,smY2019Δtg-SMT-gvpO胞外胰蛋白酶及电泳条带均与smY2019Δtg-SMT差异不大(图3-c、图3-d)。上述结果表明,表达完整的气囊蛋白基因簇相对于仅表达部分气囊结构蛋白基因能更有效的促进SMT在S.mobaraensis中的合成。
2.3 S.mobaraensis中gvp基因的转录水平分析
为确认gvp基因在S.mobaraensis中的表达情况,分别对smY2019Δtg-SMT、smY2019Δtg-SMT-gvpA、smY2019Δtg-SMT-gvpO和smY2019Δtg-SMT-gvp40587进行定量PCR分析。在smY2019Δtg-SMT-gvpA中,除了gvpA之外,gvpO、gvpS和gvpK均不同程度上调。在gsmY2019Δtg-SMT-gvpO中,gvpO、gvpG和gvpS有较明显上调(图4)。gvp基因簇各基因间的表达会互相影响已经有相关报道。例如,敲除嗜盐古菌中gvpO基因导致GvpA表达量的显著下调[8]。然而,确切gvp基因表达调控机制仍不清楚,有待后续研究。在smY2019Δtg-SMT-gvp40587中,关键调控基因gvpO和结构蛋白基因gvpA的转录水平较smY2019Δtg-SMT分别提高4.2倍和3.7倍;此外,其他gvp基因转录水平也均高于对照菌株(图4)。值得注意的是,辅助蛋白基因gvpJ仅在smY2019Δtg-SMT-gvp40587中明显上调(图4)。一般认为,gvpJ通过与gvpA相互作用提升气囊的稳定性[20]。因此,gvpJ表达水平的提升可能是smY2019Δtg-SMT-gvp40587中SMT高效合成的重要原因。
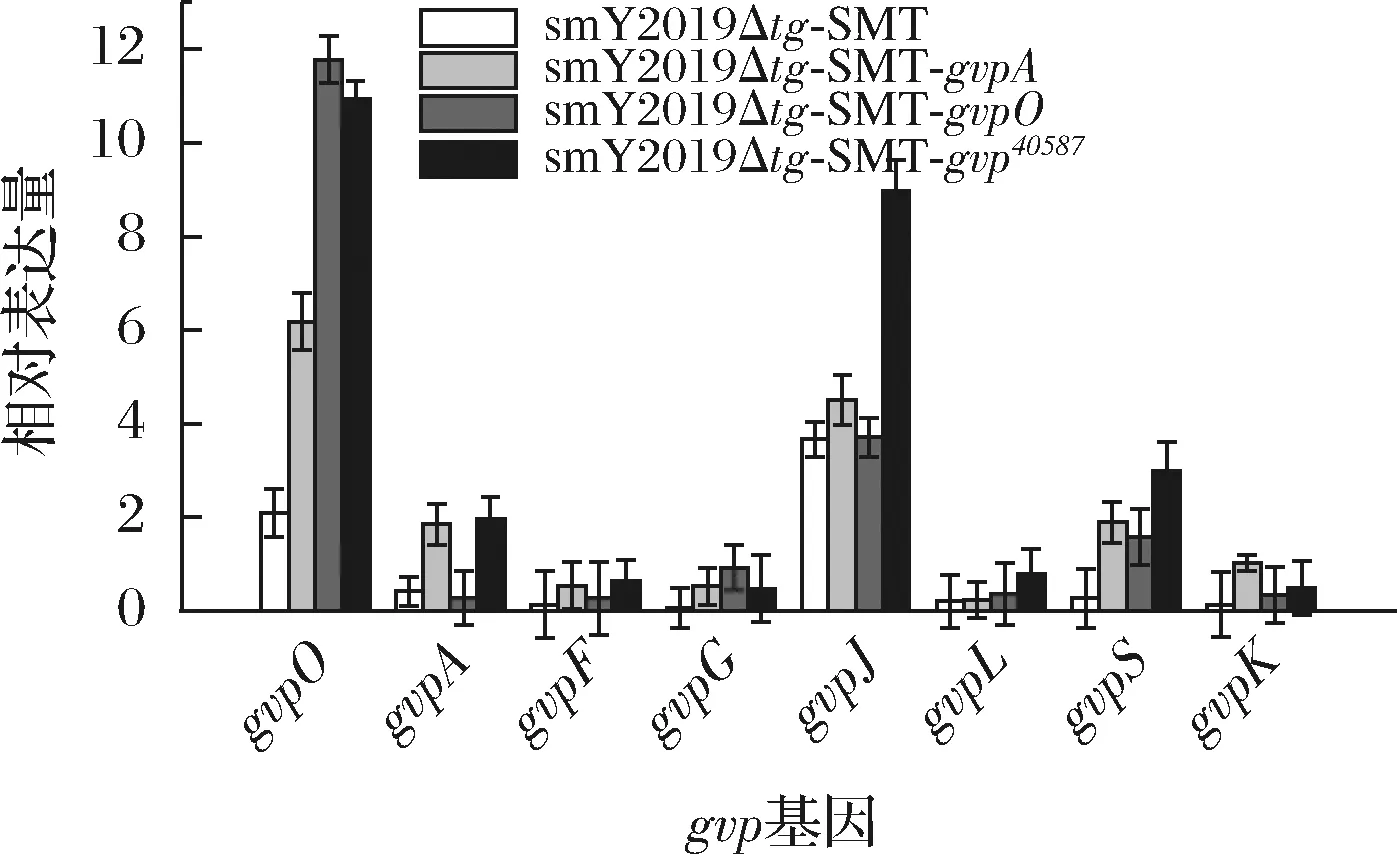
图4 S.mobaraensis中gvp基因的转录分析Fig.4 Transcriptional analysis of gvp genes in S.mobaraensis
2.4 过量表达gvp基因对S.mobaraensis生理状态的影响
由于气体囊泡内部充满气体[1],过量表达gvp基因可能有助于供氧和ATP等能量物质的合成。在Streptomycessp.CB03234-S中过量表达其内源气囊蛋白基因簇后,研究者发现了菌株的生长形态变化和次级代谢物产量增加[13],但并未对胞内的ATP等生理状态参数进行测定。为分析过量表达gvp基因对S.mobaraensis生理状态的影响,分析了gvp基因过量表达菌株的菌体生长、ATP及还原力水平等生理状态关键指标。
结果显示,smY2019Δtg-SMT-gvp40587的菌丝生长量在24~60 h略高于smY2019Δtg-SMT和其他gvp过表达菌株(图5-a)。此外,smY2019Δtg-SMT-gvp40587胞内的ATP含量和NADPH/NADP+明显高于其他菌株,且在36 h时达到最大值;其中,smY2019Δtg-SMT-gvp40587的ATP含量和NADPH/NADP+分别较smY2019Δtg-SMT高1.4倍和0.4倍(图5-b、c)。酶或蛋白质的合成依赖于胞内能量和前体供给[21]。因此,能量和还原力增加可能是SMT在smY2019Δtg-SMT-gvp40587高效合成的重要原因。值得注意的是,过量表达纤维素酶A导致了变铅青链霉菌(Streptomyceslividans)TK24中NADPH的增加[22]。目前,关于链霉菌gvp基因簇功能的研究极少,过量表达gvp基因簇影响S.mobaraensis胞内能量水平和前体供给的机制还有待进一步研究。为确认过量表达gvp基因簇是否增加了胞内气体囊泡数量,对上述重组菌进行了透射电镜分析。发酵培养基包含不溶的大豆粉,不易与S.mobaraensis菌丝分离,严重干扰了透射电镜观测。因此,选取固体培养获得的菌丝进行分析。然而,在smY2019Δtg-SMT-gvp40587和smY2019Δtg-SMT胞内均未见明显的气囊蛋白形成(图5-d),可能是由于固体培养环境不利于气体囊泡的形成。在后续研究中,将换用其他不含固形物发酵培养基发酵制备smY2019Δtg-SMT-gvp40587和smY2019Δtg-SMT菌丝体。
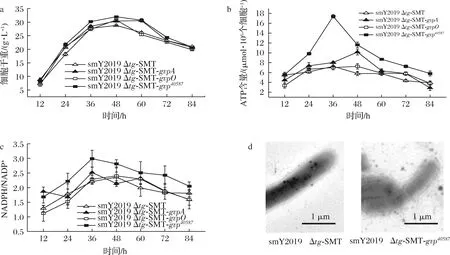
a-重组菌生长情况;b-重组菌胞内能量;c-重组菌还原力;d-透射电镜分析图5 过量表达gvp基因对S.mobaraensis生长及代谢的影响Fig.5 The effects of overexpressing gvp genes on the cell growth and metabolism of S.mobaraensis
3 结论
尽管研究者已经在大量的链霉菌中发现了gvp基因簇,然而其生理功能仍不清楚。本研究在整合表达胰蛋白酶的菌株smY2019Δtg-SMT中,分别过量表达了gvpA、gvpO以及整个gvp基因簇,首次发现了gvp基因簇或主要结构基因gvpA能促进酶在S.mobaraensis中表达。此外,过量表达gvp基因簇可显著提高胞内的ATP含量和NADPH/NADP+。因此,能量和还原力增加可能是SMT在gvp基因簇过量表达菌株中高效合成的重要原因。值得注意的是,Streptomycessp.CB03234-S和S.mobaraensis的gvp基因簇中均不含有多数链霉菌的gvpY和gvpZ,其缺失对于调控气囊蛋白形成的作用还有待进一步分析。基于gvp基因对SMT在S.mobaraensis中合成的重要影响,通过调控gvp基因的表达时间和强度可能成为提高合成蛋白及其他代谢产物产量的新策略。
