液相色谱串联质谱法同时检测消毒产品中63 种激素
2023-07-26吴芳花具笑平吴志国陈腾辉
吴芳花,具笑平,吴志国,陈腾辉
(广东省河源市药品检验所,广东 河源 517025)
消毒产品不是药品,且严格区别于药品。消毒产品无治疗疾病的作用,故成分中禁止使用激素等物料,非法添加激素可增强效果,使消费者误以为可避免药物副作用又有疗效或是直接误当药品使用,导致激素长期或过量使用,耽误病情或引发如“大头娃娃”等问题。目前,虽有研究采用液相色谱- 质谱联用法检测消毒产品,但研究覆盖的激素成分较少,检测的品种类别单一,用时较长,专属性较差,随着非法激素成分种类的增加,简单或成分单一的检测方法已难以满足监管需求,急需改进。鉴于此,本研究中以常见的63 种激素为分析目标,建立了系统、有效、快速且能在不同基质消毒产品中同时筛查和验证63 种激素成分的检测方法。现报道如下。
1 仪器与试药
1.1 仪器
Shimadzu LCMS - 8050 型液相色谱- 三重四极杆质谱联用仪(岛津<上海>实验仪器有限公司);XS205型电子天平(瑞士Mettler Toledo 公司,精度为0.1 mg);Milli - Direct16 型超纯水系统(德国Merck 公司);CT14RD型台式高速离心机(天美仪拓实验室设置<上海>有限公司);DTC-15F 型超声波清洗仪(湖北鼎泰恒胜科技设备有限公司);MX - S 型赛洛捷克涡旋混合仪(上海芃奇科学仪器有限公司)。
1.2 试药
混合激素对照品溶液A(32 种激素对照品混合,批号为S070721),混合激素对照品溶液B(30 种激素对照品混合,批号为S070383),均购自迈迪嘉<天津>科技有限公司,质量浓度均为100µg/mL;曲安西龙对照品(中国食品药品检定研究院,批号为100333-201102);N-丙基乙二胺(PSA,批号为MO0220526106),十八烷基键合硅胶(C18,批号为MO0220526110),均购自深圳逗点生物科技有限公司;无水硫酸镁(阿拉丁试剂<上海> 有限公司,批号为MO0220526110);氯化钠(批号为20201107),亚铁氰化钾溶液(批号为20161202),均购自广州化学试剂厂;乙酸锌(广东省化学试剂工程技术研究中心,批号为20170621);甲醇、乙腈均为色谱纯,水为超纯水。实测样品40 批,来自市场随机或针对性采购。
2 方法与结果
2.1 试验条件
色谱条件:色谱柱为Shim-pack XR-ODS ⅡC18柱(100 mm ×2.1 mm,2µm);流动相:水(A)-乙腈(B),梯度洗脱(0~4 min 时25%B,4~8 min 时25%B →33%B,8~10.5 min 时33%B →38%B,10.5~14.5 min时38%B →60%B,14.5~15 min 时60%B →95%B,15~17.5 min 时95%B,17.5~17.6 min 时95%B →25%B,17.6~22.5 min 时25%B);流速:0.3 mL/ min;柱温:45 ℃;进样量:3µL。
质谱条件:电喷雾离子源(ESI);正离子、负离子多反应监测模式(MRM),监测离子对及相关参数设定见表1(限于篇幅,仅选取其中较有代表性的20 种激素成分的相关参数;其中雌三醇、雌二醇采用负离子MRM,其余待测成分为正离子MRM;*离子为定量离子)。
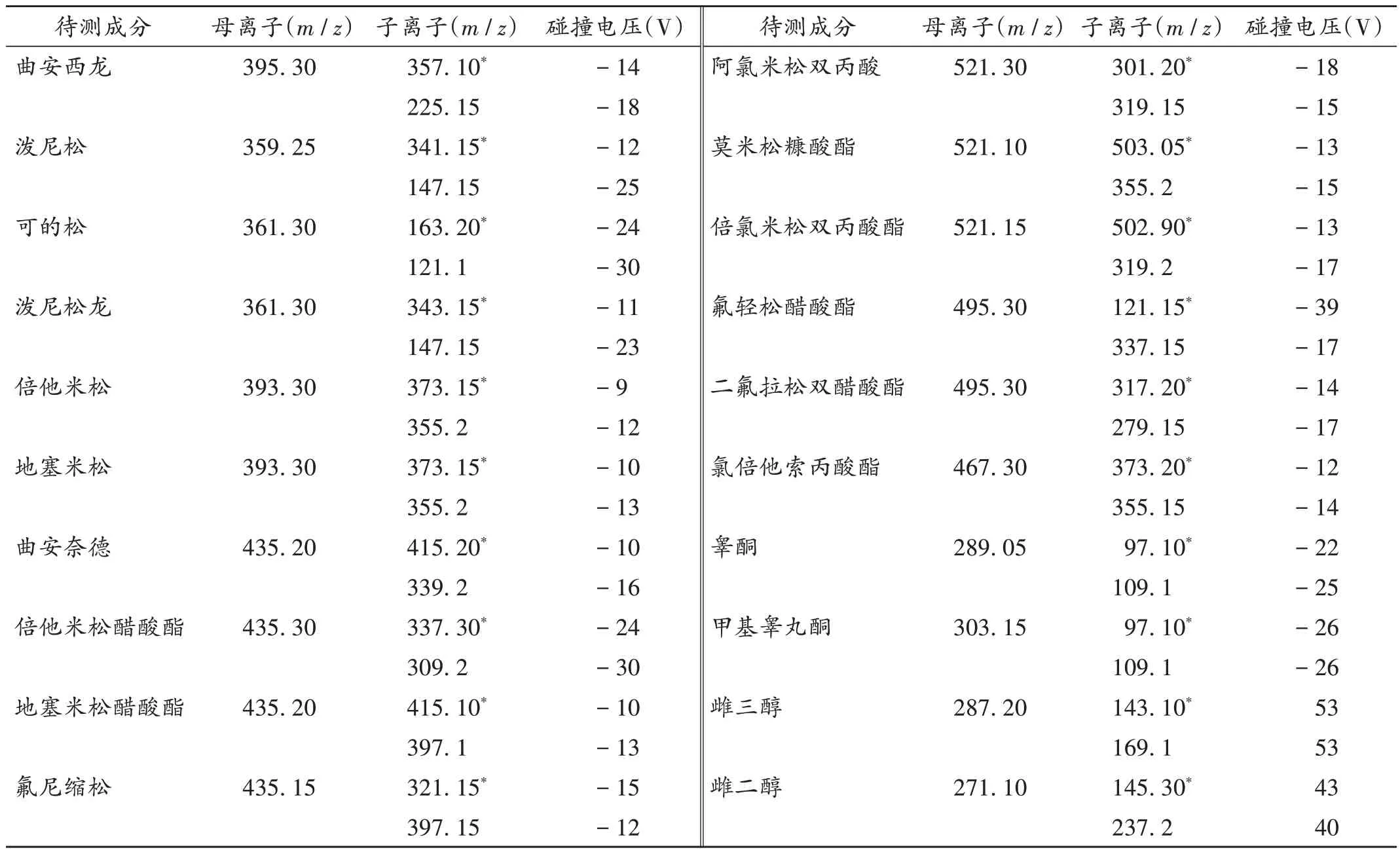
表1 激素类成分监测离子对及相关参数(部分)Tab.1 Partial monitoring ion pairs of hormones and related parameters
2.2 溶液制备
混合对照品溶液:取曲安西龙对照品约10.0 mg,精密称定,加乙腈超声(功率446 W、频率40 kHz,下同)处理20 min,制成质量浓度为100 µg/ mL 的曲安西龙对照品贮备液。精密移取混合激素对照品溶液A,B 及曲安西龙对照品贮备液各1.0 mL,置20 mL 容量瓶中,加乙腈定容,得质量浓度均为5 µg/mL 的混合对照品贮备液;精密量取5.0 mL,置25 mL容量瓶中,加乙腈定容,即得质量浓度均为1µg/mL的混合对照品溶液。
供试品溶液:1)液体制剂、凝胶制剂类。取样品(称前混匀)约0.2 g,精密称定,置10 mL 塑料离心管中,加入少量乙腈,涡旋振荡30 s 至混匀,加入乙腈至近刻度,超声提取20 min,静置至室温,用乙腈定容,摇匀,8 000 r/ min 离心10 min,滤过(当样品中地塞米松或倍他米松含量过高导致两峰难分开时,应进一步适当稀释,最后可取上清液适量加50%乙腈溶液稀释,稀释液经0.22µm滤膜滤过,即得。2)乳膏制剂类。同法取样,置15 mL 离心管中,加入2 mL 饱和氯化钠溶液,充分涡旋,精密加入乙腈5.0 mL,再次充分涡旋。超声处理15 min后混匀,于冰箱0~4 ℃冷藏1 h,然后8 000 r/min离心15 min,取上清液置15 mL 净化管中(含PSA、C18各0.2 g,无水硫酸镁0.5 g)充分涡旋3 min,同前冷藏、离心,取上清液置10 mL 离心管中,加25%乙腈2~3 mL,再加10%亚铁氰化钾溶液及20%乙酸锌溶液各0.2 mL,再加25%乙腈定容,同法冷藏,8 000 r/min离心10 min,取上清液,经0.22µm滤膜滤过,取续滤液,即得。
阴性对照品溶液及系列标准溶液:1)液体制剂、凝胶制剂类。取与待测样品基质类型相同空白样品0.2 g,共5 份,精密称定,置10 mL 离心管中,得阴性对照品溶液;分别加入混合对照品溶液适量,按供试品溶液制备方法处理,然后用50%乙腈溶液配制成质量浓度分别为10,25,50,80,100 ng/ mL 的系列标准溶液。2)乳膏制剂类。同法取样,得阴性对照品溶液。分别加入混合对照品贮备液适量,按供试品溶液制备方法处理,配制成质量浓度分别为20,40,60,80,100 ng/mL 的系列标准溶液。
2.3 方法学考察
专属性考察:取液体制剂、凝胶制剂、乳膏制剂的阴性样品各0.2 g,精密称定,置10 mL 具塞比色管中,加混合对照品溶液0.10 mL,制得相应加标溶液,滤过,取续滤液,按2.1项下试验条件进样测定,记录色谱图。结果3种加标样品溶液色谱图中,在与混合对照品溶液相同保留时间处均出现相应色谱峰,离子丰度比均在允许误差范围内,且阴性对照无干扰。详见图1。

图1 MRM色谱图Fig.1 MRM chromatograms
线性关系考察:取2.2 项下各系列标准溶液适量,按2.1 项下试验条件进样测定,记录峰面积,以待测成分质量浓度(X,ng/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。结果表明,各待测成分质量浓度,液体制剂、凝胶制剂在10~100 ng/ mL,乳膏制剂在20~100 ng/mL范围内与峰面积线性关系良好。详见表2。
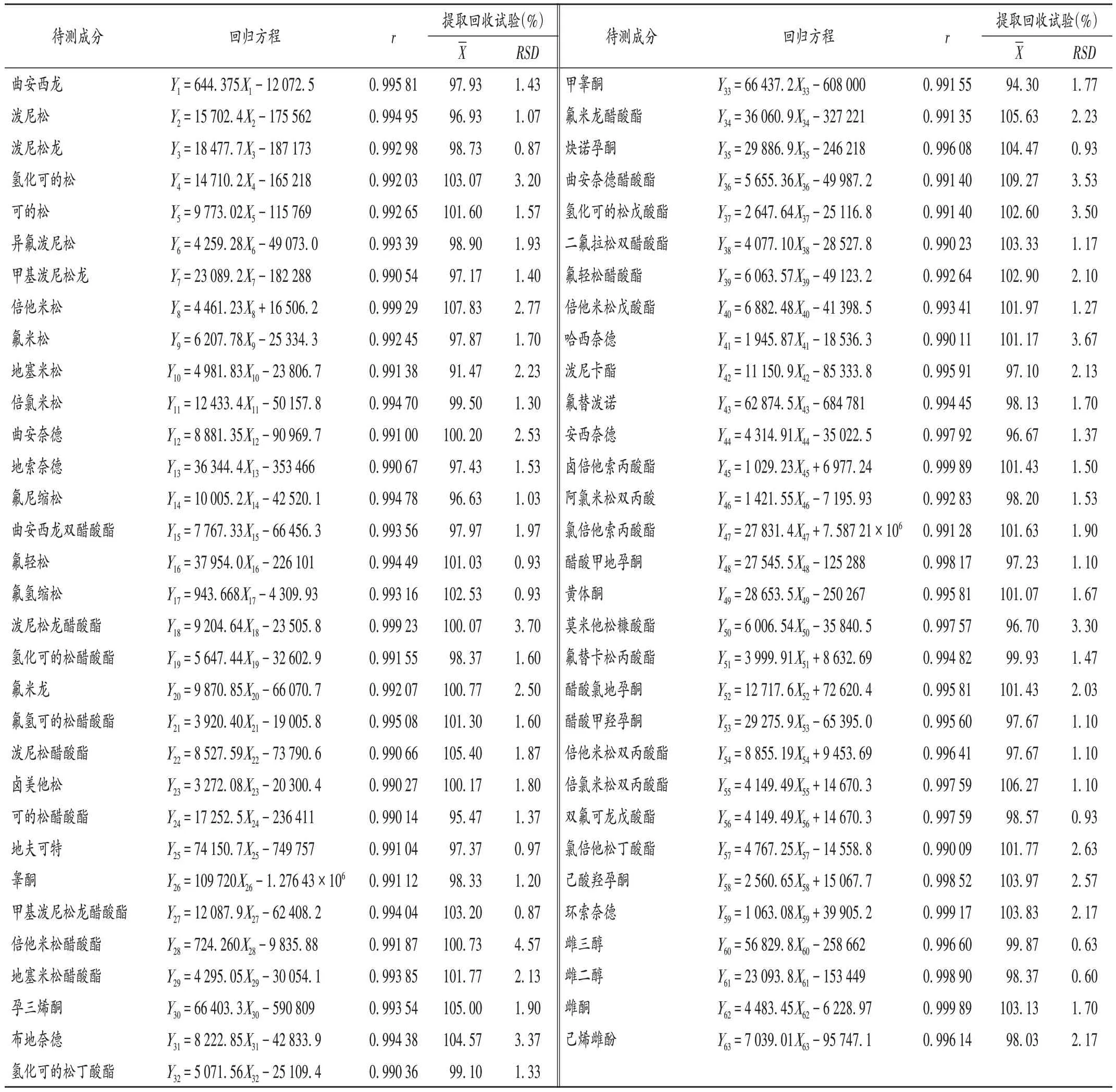
表2 方法学考察及提取回收试验结果Tab.2 Results of the methodological investigation and extraction recovery test
检测限与定量限考察:取3种空白各适量,分别添加混合对照品溶液,用50%乙腈倍比稀释,按2.1项下试验条件进样测定,以定量离子信噪比为3和10时的含量作为检测限和定量限。结果63种待测成分在液体制剂、凝胶制剂、乳膏制剂中的检测限分别为0.025~0.030µg/g,0.029~0.031 µg/ g,0.028~0.033 µg/ g,定量限分别为0.078~0.110 µg/ g,0.080~0.123 µg/ g,0.083~0.124µg/g。
精密度试验:精密吸取混合对照品溶液0.5 mL,置10 mL 容量瓶中,加50%乙腈定容,制成质量浓度为50 ng/ mL 的混合对照品溶液。按2.1 项下试验条件连续进样测定6 次,记录峰面积。结果63 种激素定量离子对质谱峰峰面积的RSD为0.414%~6.622%(n=6),表明仪器精密度良好。
稳定性试验:取3 种不同基质阴性样品适量,分别于室温下放置0,1,2,4,8,12,24 h时按2.1项下试验条件进样测定,记录峰面积。结果的RSD为0.52%~10.23%(n=7),表明室温下上述阴性样品放置24 h 内稳定性良好。
重复性试验:分别取稳定性试验项下供试品溶液各6 份,分别加入混合对照品溶液,混匀,按2.2 项下方法制备供试品溶液,按2.1 项下试验条件进样测定,记录峰面积。结果液体制剂的阴性样品峰面积的RSD为3.82%~13.51%,凝胶制剂的为2.31%~13.22%,乳膏制剂的为5.20%~10.02%(n= 6),表明方法重复性良好。
提取回收试验:取3 种不同基质阴性样品各适量,加入适量混合对照品溶液,混匀,按2.2 项下方法制备供试品溶液,按2.1 项下试验条件进样测定,计算提取回收率。结果见表2(因篇幅有限,仅列举液体制剂)。
2.4 样品含量测定
取40 批样品各适量,按2.2 项下方法制备供试品溶液,按2.1 项下试验条件进样测定,以外标法计算非法添加激素的含量。结果12 批(30.00%)样品检出非法添加激素成分。其中2批同时添加地塞米松及地塞米松醋酸酯,含量分别为0.04 mg/g、0.06 mg/g及0.59 mg/g、0.83 mg/ g;8 批添加氯倍他索丙酸酯,含量分别为0.58,0.82,0.06,0.10,1.25,4.56,3.12,3.60 mg / g;2 批同时添加地塞米松、地塞米松醋酸酯、氯倍他索丙酸酯,含量分别为0.007 mg/g、3.07 mg/g、0.004 mg/g及0.005 mg/g、2.43 mg/g、0.002 mg/g。
3 讨论
3.1 样品提取方法优化
预试验中采用实验室常用试剂(如甲醇、乙醇、乙腈等)作为提取液,乙腈溶解性强,回收率较高,因此选择乙腈作为液体和凝胶制剂样品的提取溶剂。而乳膏类样品由于基质中含有大量的油脂、糖和表面活性物质,单纯用乙腈提取虽能提取出激素类成分,但重复性欠佳,尤其是有机相类,能满足单纯的定性要求,但难以精确定量。通过QuEChERS 提取技术[1-4]+ 亚铁氰化钾溶液- 乙酸锌沉淀法净化样品,并通过各种比例和两项技术顺序调整进行探索,最终形成本研究中的提取方法,回收率和重复性大幅提高。同时,通过预试验研究加入亚铁氰化钾溶液- 乙酸锌溶液和QuEChERS系统净化的先后顺序,确定最后加亚铁氰化钾溶液-乙酸锌溶液,使白色絮状物在冷冻离心后能沉淀在试管底部,样品溶液取样不受影响,能精准定量,有效解决某些样品在加入原始流动相后产生白色絮状物且冷冻离心仍无法去除最终影响定量结果的问题。
3.2 样品基质效应
消毒产品基质较复杂,尤其是乳膏制剂,与分析物一起流出的组分直接影响电喷雾离子化效果并可增强或减弱检测信号,为消除基质效应对待测成分测定的影响,在多次试验,并查阅文献[4-16]及国家药品监督管理局《化妆品安全技术规范(2015 版)》后,采用补偿基质效应的方法:以基质标准溶液加相同基质空白样品用与样品相同的前处理方法得到的供试液,用于对检测结果的校正。试验结果表明,该方法有效解决了本类样品基质干扰定量分析,特别是含量极低的待测成分含量检测的准确性问题,为应对日后复杂多变的样品基质干扰检测结果提供了良好的解决方案。
3.3 与传统方法比较
本研究中采用简单试剂和直接提取的方法,即可对各种不同基质样品同时进行63种激素成分的定性和定量检测;同时通过将流动相由原来常用的甲醇改为乙腈,使待测成分中的同分异构体能较好地分离,尤其对一直难以分离的地塞米松与倍他米松有显著效果。相对传统操作烦琐的化学反应、薄层色谱、高效液相色谱反复验证分析法,很好地节约了检测成本,节省了检测时间;同时也为检测各种非法添加药物提供了思路和良好的鉴定方法。
