男性卡尔曼综合征合并先天性心脏病1例报告并文献复习▲
2023-07-17叶丽姿阳焕军周圣明
叶丽姿 阳焕军 周圣明
(孝感市第一人民医院内分泌科,湖北省孝感市 432100)
卡尔曼综合征是伴有嗅觉缺失或减退的特发性低促性腺激素性性腺功能减退症(idiopathic hypogonadotropic hypogonadism,IHH),还可能合并其他非生殖相关的临床异质性表现。卡尔曼综合征在临床上较为罕见,IHH总体发病率为1/100 000~10/100 000,而50%~60%的IHH为卡尔曼综合征患者,男性发病率约为女性的5倍,以散发为主,少数呈现“家族性”特点[1]。卡尔曼综合征主要导致患者第二性征不发育,引发不孕不育和重大的社会心理学问题,以及性激素缺乏所致的内分泌代谢异常。因此,本文报告我院内分泌科收治的1例合并先天性心脏病非生殖相关临床异质性表现的卡尔曼综合征患者的诊治过程,并复习国内外相关文献,旨在提高临床医师对该病的认识。
1 临床资料
患者男性,53岁,因“性发育迟缓及嗅觉丧失50余年,腰背部疼痛半月余”于2020年9月7日入住我院内分泌科。患者自诉自小性器官发育较同龄人迟缓,嗅觉丧失,外生殖器呈幼稚型,如同1岁小儿;无胡须、腋毛及阴毛生长,无喉结发育,无阴茎勃起及性功能,未婚未育,声音尖细,未予以重视及特殊诊治。近半月腰背部疼痛明显,为求进一步诊治,特来我院就诊。既往史:2018年7月20日至2020年4月16日患者多次因胸闷于孝感市中心医院心内科住院治疗。心脏超声多普勒提示室间隔膜部瘤(基底部宽约1.69 cm、直径约1.1 cm),房间隔膨胀瘤(基底部宽约2.0 cm、直径约1.17 cm),升主动脉增宽4.0 cm;冠状动脉及主动脉造影检查提示左前降支近段至远段长条囊状扩张,回旋支近段至远段长条囊状扩张,其余冠状动脉未见扩张或狭窄。故诊断为:(1)冠状动脉扩张症;(2)先天性心脏病,房间隔膨胀瘤,室间隔膜部瘤。患者拒绝手术治疗,暂时予以拜阿司匹林肠溶片抗血小板聚集及盐酸地尔硫卓预防血管痉挛等药物保守治疗。个人史及家族史:父母近亲结婚并育有两子一女,父母及姐姐在其儿童时期已故,死因及发育情况不详,弟弟发育正常,已婚并育有一子一女,子女均发育正常。入院体格检查:生命体征平稳,身高180.1 cm,体重78.1 kg,体质指数24.6 kg/m2,指间距187 cm,上部量86 cm,下部量94.1 cm;面白无须,无喉结,声音尖细,无阴毛及腋毛,阴茎长约2.0 cm,双侧睾丸未触及,呈幼稚型,Tanner分期1期(见图1),无男性乳房女性化,心、肺、腹部检查未见明显异常。主要实验室及影像学检查:(1)性激素检查(2020年9月8日),雌二醇水平<25.0 pg/mL(参考范围为<75 pg/mL,下同),泌乳素水平为6.70 ng/mL(3.6~16.3 ng/mL),卵泡刺激素水平为1.70 mIU/mL(2.1~18.6 mIU/mL),黄体生成素水平为<0.2 mIU/mL(1.7~11.2 mIU/mL),睾酮水平为14.2 ng/dL(262~870 ng/dL),孕酮水平为0.18 ng/mL(0~0.46 ng/mL)。(2)其他相关检查(2020年9月8日),生长激素水平为0.064 ng/mL(0.033~2.47 ng/mL),促肾上腺皮质激素水平为22.45 pg/mL(7.2~63.3 pg/mL),甲状腺功能、皮质醇、血常规及其他生化检查均未见明显异常。(3)染色体核型测定(2019年12月31日),外周血染色体核型为46,XY。(4)嗅区及垂体磁共振(2019年12月31日),双侧嗅球体积缩小,左侧嗅束未见,双侧嗅沟变浅,垂体饱满(见图2),考虑卡尔曼综合征,建议结合临床及实验室检查。(5)脊柱全平片(2020年9月8日),胸、腰椎骨质增生。(6)肝、胆、胰、脾、肾、输尿管彩色多普勒超声检查(以下简称彩超)(2020年9月9日),肝、胆、胰、脾、输尿管未见明显异常,双肾多发囊肿。(7)睾丸及附睾彩超:2020年9月9日行第一次睾丸及附睾彩超未见睾丸亦未提示隐睾,2020年9月11日上级医师复查睾丸及附睾彩超提示隐睾可能(见图3)。(8)腰椎双能X线检查(2020年9月9日),T值为-3.7,提示骨质疏松。
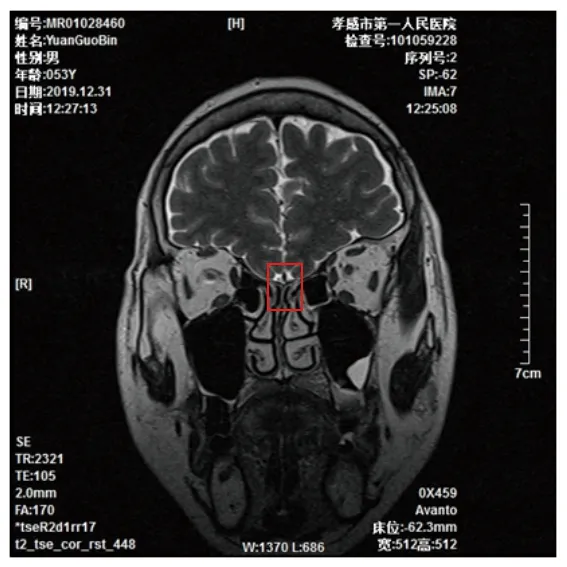
图2 患者嗅区磁共振结果

图3 患者阴囊睾丸腹股沟彩超结果
注:A左侧腹股沟可见13.8 mm×13.0 mm稍低回声团;B右侧腹股沟可见15.9 mm×14.8 mm稍低回声团。
初步诊断:(1)卡尔曼综合征;(2)先天性心脏病、房间隔膨胀瘤、室间隔膜部瘤;(3)继发性骨质疏松症;(4)冠状动脉扩张症;(5)双肾囊肿。2020年9月10日开始口服雄激素十一酸睾酮胶丸(N.V.Organon,国药准字J20100060,40 mg/次, 2次/d);抗骨质疏松治疗:晨起口服阿伦磷酸钠片(杭州默沙东制药有限公司,国药准字J20130085,70 mg/次,1次/周),口服骨化三醇胶丸(上海罗氏制药有限公司,国药准字J20100056,25 μg/次,2次/d),口服碳酸钙D3片(惠氏制药有限公司,国药准字H10950029,1片/次,2次/d)等。治疗7 d后,患者精神、体力及腰背部疼痛较前稍好转,建议其完善卡尔曼综合征相关基因检测,患者拒绝。病情好转后于2020年9月17日出院,出院后坚持服用上述药物。随访至今,患者未诉明显腰背部疼痛,精神、体力较前有所好转。
2 讨 论
卡尔曼综合征是一种具有临床表型异质性和遗传异质性的内分泌系统罕见病,除嗅觉减退或缺失、性发育迟缓这两大基本特征外,还可合并双手连带运动、眼球运动异常、听力下降、肾脏发育不全、肾上腺发育异常、先天性心脏病、智力低下、唇腭裂、牙釉质发育不全、小脑共济失调等非生殖相关的临床异质性表现,需要对患者进行完整的体格检查[2]。本例患者自幼嗅觉缺失,第二性征发育迟缓,符合典型的卡尔曼综合征临床表现,还具有房间隔膨胀瘤、室间隔膜部瘤等先天性心脏病的异质性表现,与上述研究结果相似[2]。同时,该病患者容易出现社会心理学方面的异常,临床工作中应多关注患者的心理健康,询问病情及体格检查时应尽量避开其他人员,必要时进行一对一心理咨询[3]。
卡尔曼综合征目前已知的遗传方式主要有 X-连锁隐性遗传、常染色体显性遗传、常染色体隐性遗传,其发病机制尚不明确,可能与基因突变导致胚胎早期促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)神经元和嗅神经元发育缺陷或共享的神经迁移通路出现障碍有关。胚胎发育过程中,起源于外胚层嗅基板的GnRH神经元与嗅神经纤维形成含有GnRH的轴突束,其通过筛板到达发育中的嗅球,然后通过前脑到达下丘脑,如果神经元的发育或者迁移过程发生障碍会出现下丘脑GnRH分泌缺陷及嗅神经萎缩。GnRH分泌缺陷会导致患者幼年时出现小阴茎、隐睾、第二性征发育迟缓、性功能障碍等IHH症状,嗅神经萎缩则使患者出现嗅觉减退或者缺失[4]。目前已知的与卡尔曼综合征相关的基因主要有KAL1、WDR11、NDNF、NSMF、FGFR1、FGF8、FGF17、IL17RD、PROK2、PROKR2、HS6ST1、CHD7、SEMA3A、TUBB3、SOX10等,30%的卡尔曼综合征病例由上述基因突变所致[5-6]。这些突变基因往往与某些特定的非生殖相关临床表型相关联,针对这一临床特征进行特定基因的筛查有助于发现及诊断卡尔曼综合征的突变基因。例如KAL1基因主要与镜像运动、单肾发育不全相关,FGF8基因、FGFR1基因主要与唇腭裂、牙齿发育不全、指/趾畸形等相关,PROK2基因、PROKR2基因主要与肥胖、睡眠障碍相关,CHD7、SOX10、IL17RD基因主要与听力障碍、耳聋相关[7]。WDR11基因是由Kim研究小组于2010年发现的卡尔曼综合征的致病基因之一,其可以与编码嗅神经元发育中的同源转录因子EMX1基因相互作用,导致小鼠出现卡尔曼综合征[8]; Kim等[9]发现,敲除WDR11基因的纯合子小鼠发生右心室双出口、室间隔缺损等先天性心脏病的风险高达31%(5/16),进一步揭示了WDR11基因突变可能与卡尔曼综合征患者非生殖相关的先天性心脏病表型相关联。本例患者合并有房间隔膨胀瘤、室间隔膜部瘤等先天性心脏病的异质性表现,这可能与WDR11基因突变有关,建议患者进一步完善基因检测,患者因经济原因拒绝。
卡尔曼综合征需完善较复杂的实验室及影像学检查来明确诊断。GnRH神经元发育或迁移障碍导致下丘脑-垂体-性腺轴发育异常,垂体产生的卵泡刺激素、黄体生成素减少,随之睾丸、卵巢产生的睾酮、雌激素等亦减少,但很少累及泌乳素、生长激素、甲状腺激素、糖皮质激素等其他内分泌激素分泌,性激素及垂体激素的测定可用于排除有无其他下丘脑、垂体病变引起的继发性IHH[7]。雌激素、睾酮水平的持续低下常导致继发性骨质疏松,完善双能X线检查可明确是否合并骨质疏松症[10]。卡尔曼综合征患者染色体核型正常,可与克兰费尔特综合征、特纳综合征等染色体异常引起的生殖器官发育异常相鉴别,但因其可出现性腺发育异常或合并非生殖相关的临床异质性表现,需完善睾丸、子宫、卵巢、心肾等超声检查明确有无器官发育异常,这也可为卡尔曼综合征表型相联系的基因型提供一定线索。嗅脑磁共振检查可发现患者嗅球、嗅沟、嗅束的发育异常,为诊断该疾病提供解剖学基础,同时还可排除有无其他颅脑疾病。本例患者内分泌激素检查示卵泡刺激素、黄体生成素、睾酮水平低,泌乳素、生长激素、促肾上腺皮质激素、皮质醇、甲状腺激素等水平正常,染色体核型正常,双能X线提示骨质疏松,超声检查示睾丸、心脏发育异常,颅脑磁共振示左侧嗅束未发育、双侧嗅沟发育不良、双侧嗅球萎缩,卡尔曼综合征临床诊断明确。患者第一次完善睾丸及附睾彩超提示未见睾丸及隐睾,但结合卡尔曼综合征的临床表现,不排除性腺未发育或者妊娠期、出生前后睾丸扭转血管栓塞致睾丸萎缩的可能,考虑隐睾可能性大,故再次复查睾丸及附睾彩超,提示阴囊未见睾丸回声,但两侧腹股沟区可见稍低回声团,考虑隐睾可能。建议患者进一步行人绒毛膜促性腺激素(human chorionic gonadotrophin,hCG)刺激试验明确是否存在睾丸间质细胞。此外,患者可行GnRH兴奋试验以明确下丘脑-垂体-性腺轴是否启动。但由于我院为基层医院,尚未开展GnRH兴奋试验和hCG刺激试验,故本病例缺少上述两项检查结果,患者今后可至上级医院完善相关检查。
早期诊断和早期治疗是改善卡尔曼综合征预后的关键因素。患者婴幼儿时期缺乏性征发育表型,往往难以鉴别疑似患儿,只有极少数患者在新生儿期因小阴茎、隐睾就诊,大部分患者在青春期晚期或者成年早期因典型的临床表现及精确的实验室检查方可明确诊断。因此,大部分卡尔曼综合征患者的诊断及治疗相对滞后,预后相对较差。作为一种基因突变疾病,建立敏感、快速的基因诊断方法用于产前筛查、早期诊断已成为未来防治卡尔曼综合征的重要内容,但由于卡尔曼综合征致病基因的寡聚性,目前尚未建立针对卡尔曼综合征的有效的早期基因诊断手段[11]。微小青春期的存在使得早期诊断卡尔曼综合征成为可能。生长发育过程中GnRH的分泌有两个高峰期,分别是新生儿期的微小青春期及青少年时期的青春期。微小青春期,新生儿的下丘脑-垂体-性腺轴处于兴奋状态,使得性激素水平往往高于幼儿期及儿童期,甚至接近青少年时期的正常低值,这是早期诊断卡尔曼综合征的最佳窗口时期,男孩窗口期通常在出生后至6个月,女孩通常在出生后至2年[12]。有卡尔曼综合征家族史或者有性器官发育异常的新生儿,可早期测定窗口期性激素水平,尽早诊断或排除卡尔曼综合征可能。
卡尔曼综合征的病理生理特征决定其外周性腺组织仍保留一定的生殖功能,若早期给予激素替代治疗则可促进其第二性征的发育,采用促性腺激素或GnRH治疗甚至可使部分男性患者产生精子,诱发女性患者排卵,从而获得生育能力[13]。作为一种基因突变疾病,通过靶向基因治疗,可以纠正人体错误的基因信息从而根治该病。然而,目前尚缺乏针对基因突变的对因治疗,相信随着分子生物学的飞速发展,卡尔曼综合征的产前筛查、早期诊断在未来有望实现突破[11]。嗅觉障碍、肾脏发育不全等非生殖表型和性激素缺乏的生殖表型是卡尔曼综合征的主要临床表现,目前针对合并嗅觉障碍、肾脏发育不全、双手连带运动等的患者暂无特殊治疗方法,针对合并唇腭裂、听力下降、骨骼发育异常、先天性心脏病等的患者需要尽早开展手术或者专科干预,针对性激素缺乏的患者则讲究个体化治疗原则,根据患者不同的年龄段、对生育的要求及垂体功能状态采取不同的治疗方法,使患者恢复性功能,尽可能地恢复生育能力[14]。
卡尔曼综合征较为理想的治疗方案是尽量模拟正常的青春期生长发育过程。目前,男性卡尔曼综合征的治疗方案主要有雄激素替代疗法、促性腺激素生精疗法、GnRH泵生精疗法。男性患者在婴幼儿期及儿童期的治疗重点在于使睾丸下降、阴茎生长。隐睾症对患儿未来的生育能力影响较大,建议尽早手术矫正[15];微阴茎患儿应早期采取低剂量的短期性激素疗法,诱导患儿阴茎发育至接近同龄人,减轻患儿及家长的心理负担,并注意监测骨龄[16]。在患儿青春期及成年期,应使用睾丸激素促进其第二性征发育,维持骨骼健康,消除其对未来生育能力的担忧,保持心理健康。对于有生育要求的患者,一旦达到男性化目的,则可开始刺激精子发生、成熟,GnRH泵或者hCG联合人绝经期促性腺激素双促生精方案均可刺激睾丸间质细胞产生睾酮、睾丸生殖细胞的增殖和成熟,诱导生精管生精[17]。当睾丸初始体积<4 mL时,可采用GnRH泵或促性腺激素联合治疗,国外已有研究结果表明,上述两种疗法的生精效果相当,但国内有研究报告,脉冲式GnRH泵疗效优于促性腺激素联合治疗[18]。朱大龙等[19]开展的针对男性IHH的临床研究显示,人hCG联合间断尿卵泡刺激素序贯疗法对男性IHH患者性腺发育、精子生成的作用并不劣于传统序贯疗法,但注射次数及注射费用显著降低,可作为治疗男性IHH的双促方案。对于睾丸初始体积≥4 mL且无隐睾史的患者,可先选用hCG单一疗法诱导精子发生,若治疗3~6个月后射精中仍无精子,可采用人绝经期促性腺激素刺激曲细精管生精[18]。对于精子质量或数量严重受损的卡尔曼综合征患者,经上述常规治疗后效果欠佳时,可联合体外受精等辅助生殖技术来提高生育能力。本例患者未婚未育,50岁后才正规就诊,已错过最佳诊疗时机,且合并有先天性心脏病、严重的骨质疏松等并发症,患者拒绝心脏手术治疗,暂时予以补充雄激素、抗骨质疏松等治疗,电话随访5个月患者诉精神、体力、胸背部闷痛等均好转。
3 小 结
卡尔曼综合征是一种同时具备嗅觉障碍、先天性性腺功能减退两个特征的罕见异质性疾病,还可合并先天性心脏病、肾脏发育不全、双手连带运动等其他非生殖相关表型。早期诊断和早期治疗是改善卡尔曼综合征预后的关键因素。有卡尔曼综合征家族史或者性腺发育异常的患儿,应高度警惕卡尔曼综合征可能,尽早完善性激素测定、嗅区磁共振、基因检测等检查明确或排除诊断。应根据患者的年龄、生育要求、垂体功能状态等选择激素替代治疗方案,以达到维持患者第二性征发育、最大限度恢复患者生育能力的目的,而针对合并严重先天性心脏病的患者则应考虑手术治疗。
