基于SNP和表型性状的籼稻种质资源遗传多样性研究
2023-07-15贺乔乔周希希王业文李培江王胜宝
贺乔乔 周希希 王业文 李培江 王胜宝 张 羽*
(1.陕西理工大学 生物科学与工程学院/陕西省资源生物重点实验室,陕西 汉中 723000; 2.陕西省水稻研究所,陕西 汉中 723000)
水稻(OryzasativaL.)是全球主要的粮食作物,也是植物遗传育种和基因组学研究的模式生物,在种间、种内都具有丰富的遗传多样性。种质资源评价可以为加强种质资源的管理、保护与利用奠定基础。由于不同水稻材料在每个农艺性状上表现出丰富的表型多样性,胡标林等[1]以14个表型性状对1 579份全球水稻种质资源进行遗传多样性和优良稻种的分析与评价,结果表明株高、抽穗期、倒伏性、淀粉含量、颖壳色和糙米色性状可作为种质资源综合评价的指标。张晓丽等[2]采用统计数据和主成分分析法,对4个东南亚国家的298份水稻种质资源进行表型多样性分析,结果表明有效穗数的变异系数最大,株高、茎秆长和穗粒数次之,结实率的变异系数最小。23个表型性状中前8个主成分在总变异中的累计贡献率达67.99%。张雯雯等[3]用18个农艺性状对56份云南省滇西北地区的粳稻资源的表型多样性进行分析,结果表明数量性状的多样性指数(1.68~2.06)明显高于质量性状(0.60~1.16)。汤翠凤等[4]用17个农艺性状的多样性指数对云南省1 189份水稻地方品种进行了表型多样性鉴定。
随着分子生物学的不断发展,分子标记技术在生物学各个领域的应用已日趋成熟。特别是高通量测序技术的发展是获得第三代分子标记SNP(single nucleotide polymorphism)的有效途径,SNP具有在基因组分布密度高、多态性高、遗传稳定和检测方法众多等特点[5],能最大化利用DNA序列的变化。简化基因组测序(reduced-representation genome sequencing,RRGS)是基于高通量测序技术发展起来的利用酶切降低基因组复杂程度的测序技术,主要分为限制性酶切位点DNA测序RAD-seq、基于测序的基因分型技术GBS(genotyping-by-sequencing)和特异性位点扩增片段测序SLAF-seq[6]。GBS技术的关键是运用了甲基化敏感的限制性内切酶,如NlaIII、MseI等Ⅱ型酶,回避了基因组主要的重复区域[7],简化基因组测序,具有不参考基因组便可进行大量SNP开发的优势,其在构建遗传图谱、遗传多样性、QTLs(quantitative trait locus)作图、群体遗传学等研究领域被广泛应用。刘传光等[8]报道,水稻亚种间及亚种内品种间SNPs都很丰富,在用传统的分子标记方法难以找到亲缘关系近的材料间多态性位点时,用SNPs技术也能找到数量可观的多态性位点。水稻中平均SNP的发生频率为0.65%左右。王悦星等[9]报道分布在籼稻12条染色体上的SNPs和单倍型(haplotype)的多态性及分布密度都不同。侯青青等[10]介绍了在水稻中利用SNP芯片分型和重测序SNP分型技术进行水稻重要性状全基因组关联分析的方法。李梓榕等[11]开发了一种快速筛选SNP构建DNA指纹图谱的方法,利用12个SNP标记即可对117份水稻种质进行鉴定。Singh等[12]利用SSR(simple sequence repeat)和SNP两种标记方法比较了375个印度水稻品种的遗传多样性和群体结构的差异,结果表明SNP标记具有更高的群体分辨率,但SSR对多样性分析更有效。Courtois等[13]利用25对SSR和70个SNPs标记对250份粳稻核心品种进行了表型和结构分析,结果表明SSR和SNP产生的距离矩阵相关性很强。Travis等[14]利用定制的384-SNP芯片对孟加拉的511份水稻品种的遗传结构进行分析,将511个品种划分为4个群。Choudhury等[15]利用水稻的12条染色体上的36个非连锁SNP标记,对6 984份来自印度东北部水稻材料的遗传多样性和群体结构进行分析,把6 984份水稻材料划分为3个亚群。Parida等[16]探索了水稻的抗逆性状与SNPs的相关性,在384个SNP标记中成功验证了362个标记,为鉴定和定位性状相关基因组区域提供了线索。目前,运用基因组学和表型组学对秦巴地区籼稻种质资源的遗传多样性研究鲜见报道。本研究利用SNP分子标记和15个表型性状对秦巴地区198份籼稻种质资源进行遗传多样性和群体结构分析,旨在探究籼稻表型性状的遗传多样性信息,以期为挖掘籼稻优异种质资源和分子辅助育种提供参考。
1 材料与方法
1.1 试验材料
198份籼稻种质资源包括,恢复系材料112个,保持系材料49个,恢保关系不明的材料37个,于陕西省水稻研究所试验场(106°59′57″ E,33°7′48″ N),按16.7 cm×20.0 cm分株式设计移栽(表1)。每个样品随机排列在3个地块上,地块之间没有保护边缘行。
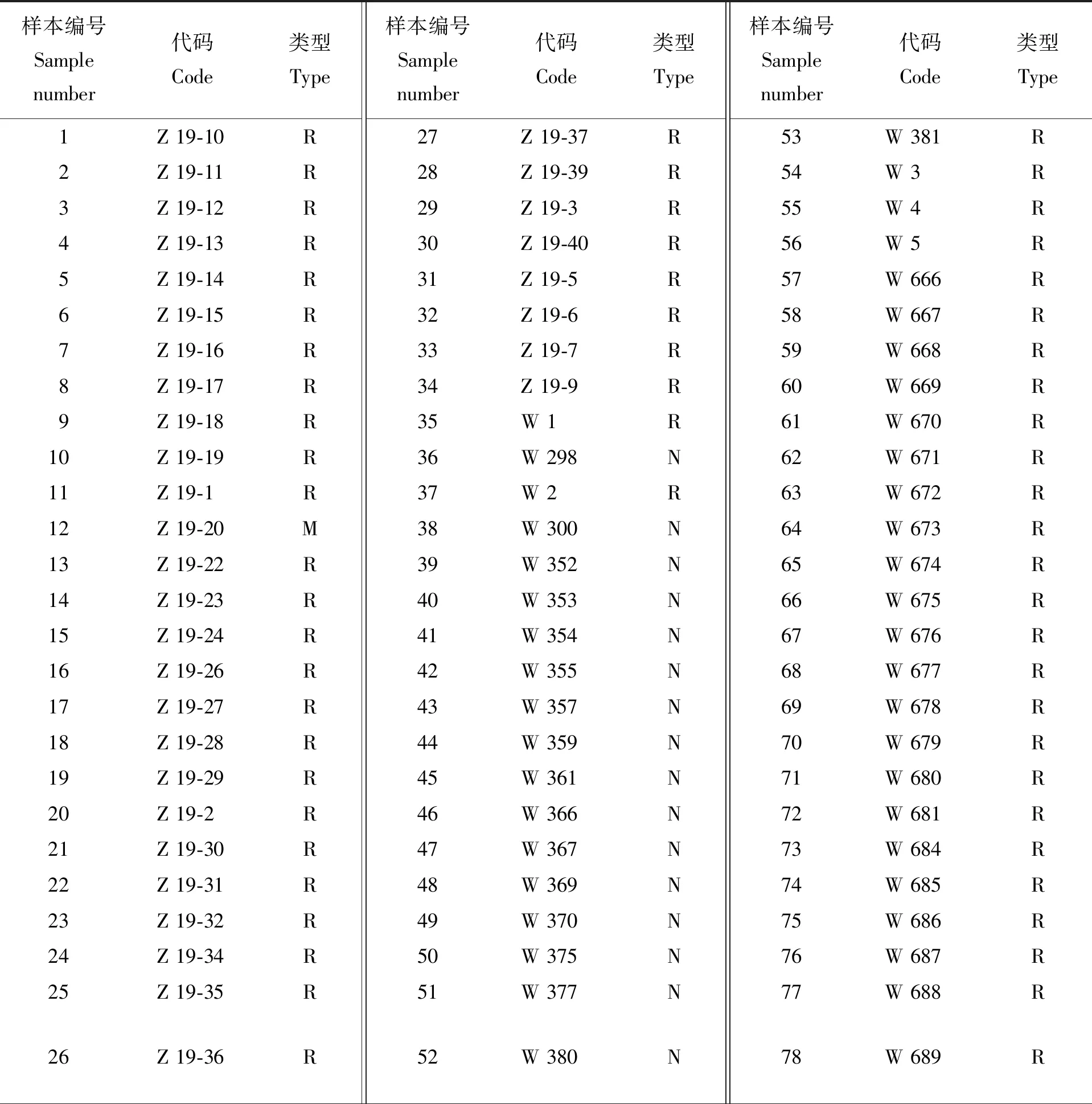
表1 供试籼稻信息
1.2 基因型数据获取
采用CTAB法[17]提取198份材料幼嫩叶片的DNA,采用NlaIII和MseI分别酶切的GBS技术测序,SNP过滤时采用MAF 0.05对基因频率进行筛选,其目的是过滤掉会影响后续群体结构分析的低质量SNP。
1.3 表型的相关指标测定
对198份材料的15个性状按照《水稻种质资源描述规范和数据标准》[18]进行农艺、经济和品质性状调查,在198个样本的区域中各选取6株植株,连续3年(2017—2019年)观察记录,以3年数据的平均值作为15个性状的表型值。15个性状包括播始历期、株高、叶长、叶宽、单株有效穗、穗长、穗总粒数、穗实粒数、千粒重、糙米率、精米率、整精米率、垩白度、垩白粒率、籽粒长宽比。
1.4 基于表型性状的遗传多样性分析

H’=-∑PilnPi[4]
式中:Pi为分析单元内某个性状第i级的材料数占总材料数的百分比。
使用SPSS 22.0软件对连续3年的表型数据进行Z-Scores标准化后,对15个表型数据进行遗传变异、相关性、主成分分析和欧式距离矩阵分析。使用MEGA 10.0(http:∥www.megasoftware.net/)软件对15个表型性状标准化后数据进行可视化聚类,一般近缘序列采用最大简约法(maxuimum parsimony,MP)或除权配对法(unweighted pair group method arithmetic mean,UPGMA),构建进化树。采用NTsys-pc v2.1软件[20]的PCA(principal component analysis)方法对主成分分析结果进行可视化处理。
1.5 基于SNPs的群体结构分析
群体结构分析与控制是进行基因关联定位的前提,因为复杂的群体结构会导致基因型和表型的假阳性关联。运用TASSEL 5.0(https:∥www.maizegenetics.net/tassel)对198个样本的SNPs进行扫描,进行基于Nei’s的遗传距离(identity-by-state,IBS)计算。采用Structure 2.3.4(http:∥taylor0.biology.ucla.edu/structureHarvesteroybase.org/tools.php)软件[21]的Bayes聚类算法,模拟群体遗传结构进行估计,设置隶属概率阈值为0.60,模拟1到5的种群K,每个K迭代5次,使用10 000个Burning,进行100 000次MCMC(markov chain monte carlo)迭代,以获得最可能的种群数量的估计。ΔK是根据K绘制的,根据Evanno等[22]提出的方法和Structure Harvester(http:∥taylor0.biology.ucla.edu/structureHarvester/)[23]平台获得的数据,确定最佳聚类数,并绘制基于模型的群体遗传结构图。使用GENALEX(https:∥biology-assets.anu.edu.au/GenAlEx/Welcome.html)进行 Mantel检验,Mantel检验是对两个距离矩阵相关性的统计方法[24]。
2 结果与分析
2.1 表型性状多样性及聚类分析
由表2可知,15个表型性状中,垩白度的变异系数(CV)最大,其CV为140.66%;糙米率CV最小,其CV为3.86%。多样性指数最高的表型性状是穗长和千粒重,其多样性指数都是2.08;最低的是垩白度,其多样性指数为1.54。基于15个表型性状的平均变异系数(CV)和平均多样性指数分别为30.33%和1.95。

表2 15个表型性状的基本统计分析和多样性
由表3可知,15个表型性状之间的相关系数(r)在-0.55~0.92。株高与播始历期、穗总粒数与穗长、穗总粒数与穗实粒数、整精米率与精米率、垩白粒率与垩白度之间均呈极显著正相关(P<0.01)。长宽比与垩白度、垩白粒率与穗实粒数、垩白度与穗总粒数、穗长与单株有效穗、叶长与垩白粒率、叶长与垩白度之间均呈显著负相关(P<0.05)。
由表4和表5可知,前3个主成分可分别解释群体变异的29.44%、16.63%、10.59%,对第一主成分贡献较大的性状包括穗长(0.76)、株高(0.75)、穗总粒数(0.70)、播始历期(0.68)、穗实粒数(0.67)、叶长(0.64)、垩白粒率(-0.61)和垩白度(-0.60),这8个性状对第一主成分的贡献值绝对值都在0.60以上,是籼稻表型性状变异的主要因素。
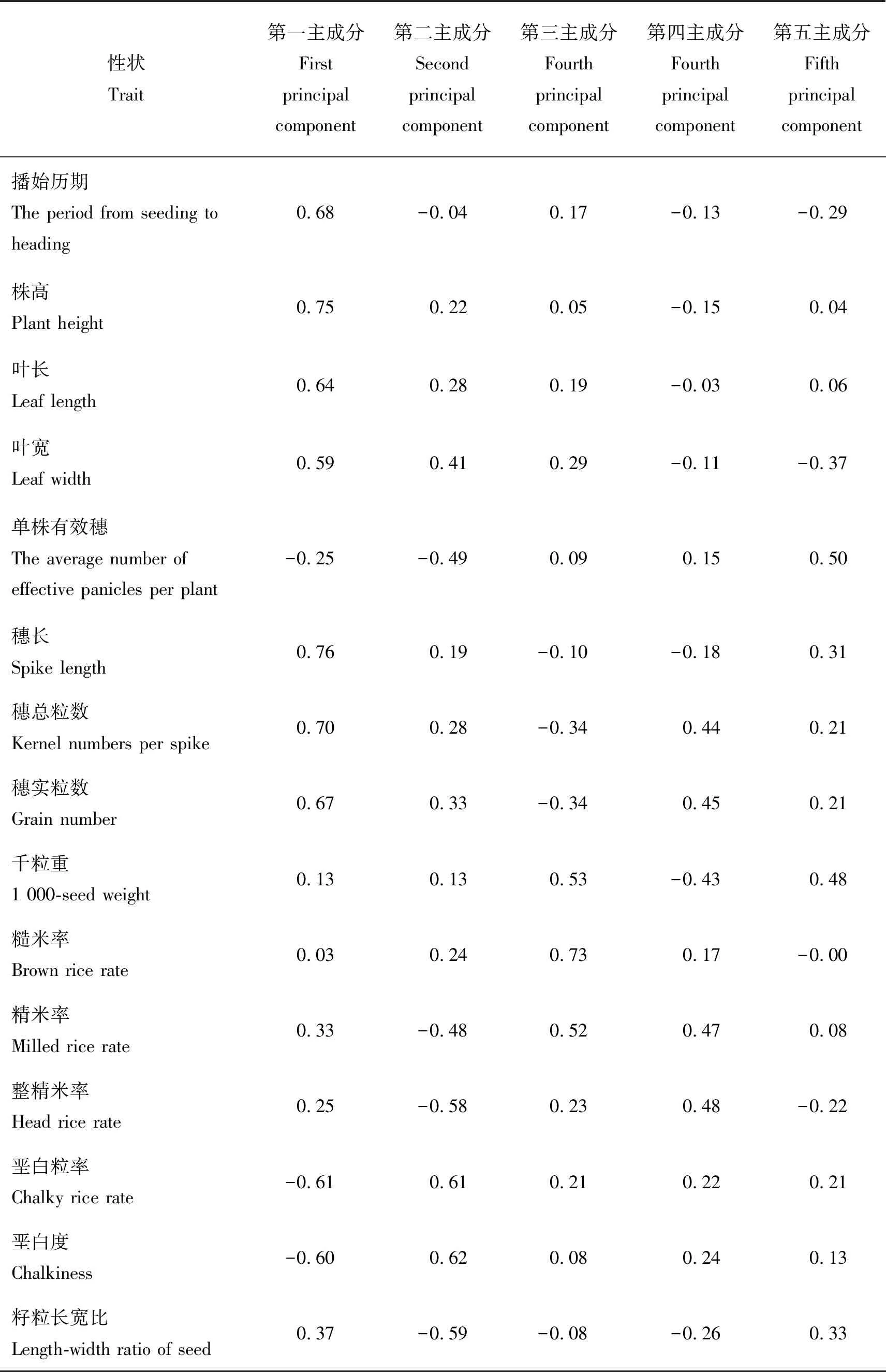
表4 15个表型性状的主成分分析的成分矩阵
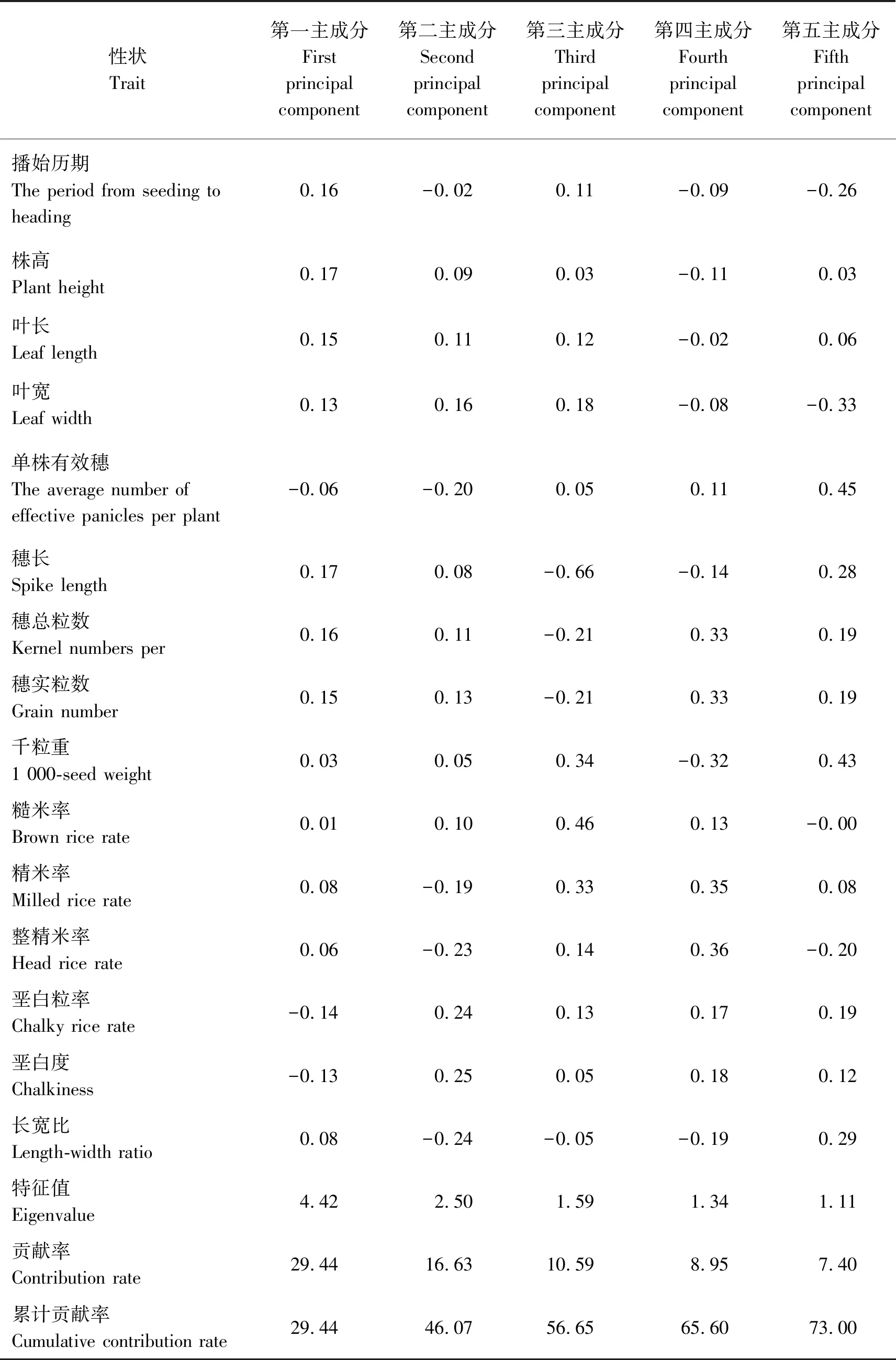
表5 15个表型性状的主成分和成分得分系数矩阵的特征值和贡献百分比
2.2 基于表型性状的聚类分析
根据标准化后的15个表型性状数据,用UPGMA聚类把198份样本聚为2个亚组(图1)。前5个主成分反映了总信息量的73.003%,前2个主成分的累计贡献率为46.072%(图2),说明198份材料的亲缘关系较近。

图1 基于15个表型性状的UPGMA聚类图
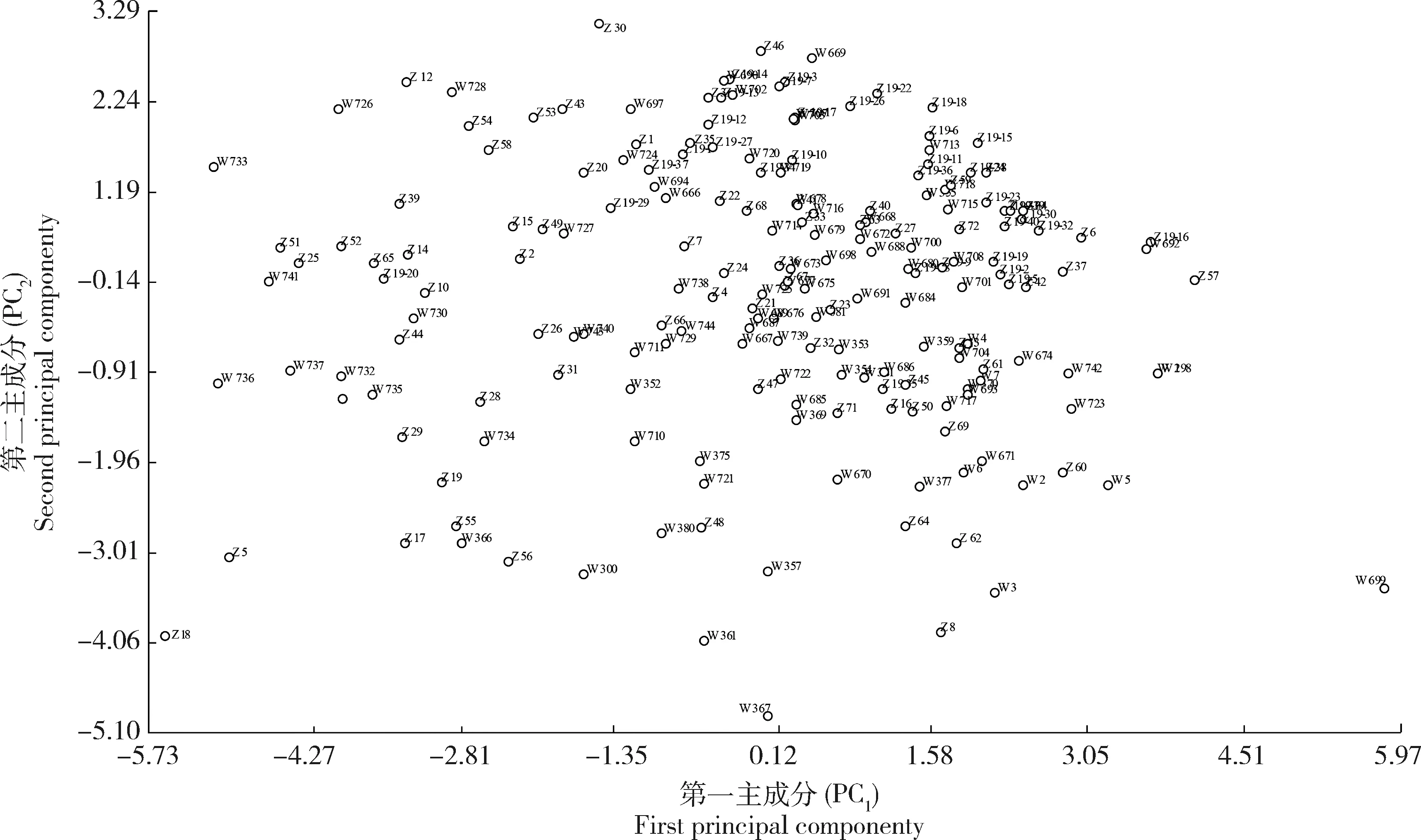
图2 基于15个表型性状的PC聚类
2.3 基因组图谱构建
分别用2种酶切的GBS技术在198份材料中共识别91 421个SNPs,其中包括85 535个比对到具体染色体上的SNPs和5 886个没有定位到具体染色体上的SNPs。杂合位点占5.85%,MAF为0.19,1~12号染色体上SNP Tajima’s D依次分别为2.006、1.815、1.182、2.097、2.087、2.271、1.732、2.003、2.057、2.499、2.274、2.064,平均值为2.007(表6)。

表6 12条染色体上SNP的Tajima’s D
2.4 基于SNPs的群体遗传结构分析
由图3可知,当K=3时,ΔK达到最大值,表明198份材料被分为3个亚类,分别由9,53,136个样本组成。基于Nei’s的遗传距离在0.014(样本Z 19-37和Z 5)~0.596(样本Z 19-37和Z 10),平均遗传距离为0.284。91 421个SNPs构成的总变异中,前3个PC可分别解释群体变异的10.98%、10.47%、4.81%,用前3个主成分聚类198份材料时分组不明显。
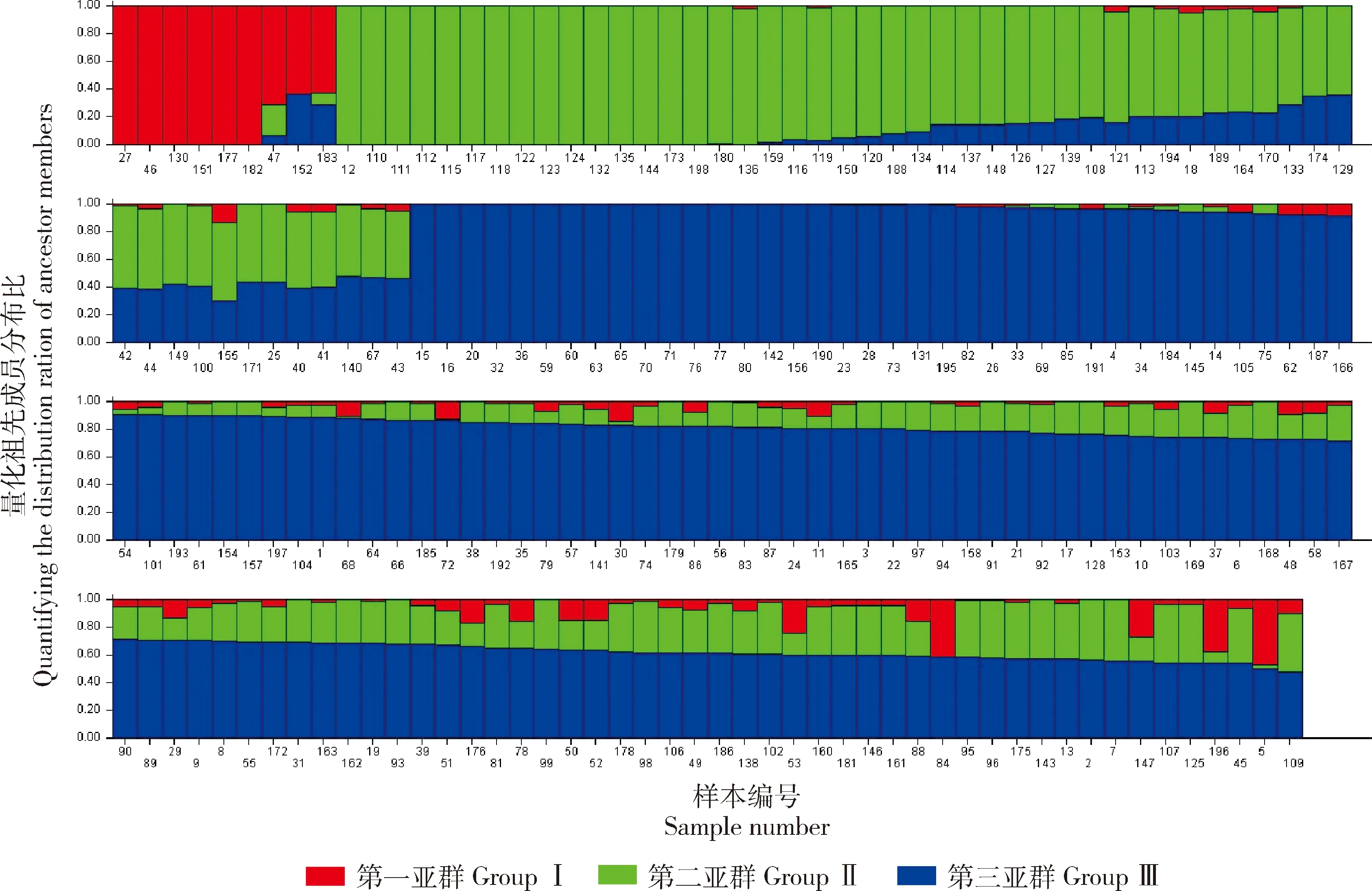
图3 K=3时,基于贝叶斯算法的遗传结构
2.5 2种聚类方法的相关性分析
Mantel检验表明,91 421个SNPs和15个表型性状的遗传距离矩阵之间的r为0.041(图4),即相关性很低,但由于表型是由基因型和环境共同决定的,表型性状中的数量性状对环境极为敏感,而在SNP分析中发现其遗传距离较近,同一育种单位育成品种遗传距离较近, 与它们的系谱来源吻合,所以认为基于SNPs的聚类比表型性状聚类更接近系谱分析。
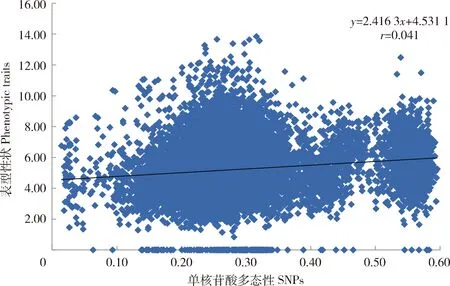
图4 基于SNPs与15个表型性状的遗传距离间的相关性
3 讨 论
水稻大部分表型性状属于数量性状遗传,如抽穗期、分蘖数、株高、每穗粒数等,这些性状对环境条件非常敏感,不同年份、不同地块的水、肥、气、热等因素均影响表型性状的表现[25]。目前大多数性状主要还是靠人工考种,容易引入主观测量误差而影响分析结果的准确性,除此之外,还可能与选择性状的数目不够多等因素有关,对表型性状的准确记录是进行全基因组基因关联定位等生物学问题分析的首要条件[26],为了保证数据的准确性,本研究进行了3次生物学重复,然而15个表型性状产生的遗传距离矩阵与SNPs标记产生的遗传距离矩阵间的相关性很低。对于不同遗传标记在群体结构和多样性的研究中,报道较多的为SSR标记与表型聚类的比较研究,张春红等[27]利用3种遗传标记(食味性状、农艺性状和SSR)对表现优异的60份粳稻材料进行了多样性分析,表明SSR标记聚类结果与系谱分析结果基本一致,而农艺性状表现与材料来源一致性较好,但与食味优良的关系不明确。赵庆勇等[28]研究发现SSR标记比表型聚类能更准确的揭示亲本之间的遗传差异。杨旺兴等[29]研究籼、粳稻及其杂交后代的遗传多样性时发现,在亲缘关系不明晰的材料中,SSR分子聚类分析比表型聚类更准确。
种质资源是作物优良基因的载体,是育种的基础,对现有资源的改良与创新利用是未来种质资源研究工作中的重中之重[30],而表型性状的应用在生物种质资源研究中起着很大的作用,随着表型组学中智能化表型测量平台的应用,表型鉴定将更加准确可靠[31],可为基因资源的发掘提供必要信息。随着测序技术的不断发展,特别是GBS技术在简化基因组测序方面的普及,越来越多的研究利用此技术开发出高密度的SNPs标记用于不同作物的资源评价、遗传图谱构建、遗传多样性分析、分子选择育种等全基因组关联研究领域[5]。SNPs标记已成功应用于水稻[32]、玉米[33]、小麦[34]、大豆[35]、葡萄[36]等作物中。本研究发现秦巴地区的籼稻种质资源的SNPs聚类比表型性状聚类更接近系谱分析,特别是亲缘关系近的材料分析时表现出强大的应用潜力,但仍需要进一步试验验证。
4 结 论
秦巴地区198份籼稻种质的SNPs构成的群体遗传结构相对简单,多种聚类分析结果表明供试的198份材料间只存在较小的遗传差异,遗传结构较为单一,而表型性状变异较丰富,多样性程度高,特别是穗长和垩白度的多样性程度高,群体间性状差异显著。穗长、株高、穗总粒数、播始历期、穗实粒数、叶长、垩白粒率和垩白度8个性状可作为秦巴地区籼稻表型性状的综合评定指标。
