羧甲司坦微囊含量测定方法的研究
2023-07-12颜元刘傲杨成刘丽娜王磊黄静
颜元 刘傲 杨成 刘丽娜 王磊 黄静
(贵州医科大学药学院,贵州 贵阳 550025)
羧甲司坦是一种粘液溶解剂,主要作用于支气管腺体的分泌,增加低粘度的唾液粘蛋白分泌,减少高粘度的岩藻粘蛋白的产生,从而降低痰液的粘稠性,同时可增加黏膜纤毛的转运,从而致使痰液易排出[1-2],目前广泛用于以痰粘液分泌过多为特征的呼吸系统疾病[3],此外羧甲司坦可通过降低Muc5ac和Muc5b的分泌,恢复Muc5b蛋白水平,从而改善患者肺功能,减轻炎症反应[4]。但由于羧甲司坦半衰期较短,当前市售的剂型(包括普通片剂、颗粒剂、溶液剂及泡腾剂)均需要一天多次给药,极大降低了患者(尤其是老年患者)的依从性和顺应性,因此将羧甲司坦制成缓释剂型不仅能减少患者服药次数,提高患者顺应性,服用方便,同时也能使其在体内缓慢平稳的释放,保持血药浓度平稳,避免出现峰谷现象,降低毒副作用及程度,从而提高患者临床用药的安全性。课题组前期已对羧甲司坦缓释片进行研究,结果表明其具有一定的缓释作用[5-6],基于此,课题组现阶段正在进行羧甲司坦微囊的研究,以期进一步提高羧甲司坦的缓释作用,在此基础上本研究以羧甲司坦微囊作为研究对象,采用紫外分光光度法和高效液相色谱法两种方法对其进行含量测定研究,建立合理、准确的含量测定方法测定羧甲司坦微囊中羧甲司坦的含量,为后续羧甲司坦微囊的处方及制备工艺筛选提供实验依据。
1 仪器与材料
1.1实验仪器 电热恒温水浴锅(山东博科再生医学有限公司,型号:DK-8D),高效液相色谱仪(岛津仪器苏州有限公司,型号:SIL-I6),紫外可见分光光度计(岛津仪器(苏州)有限公司),型号:UV-2700)),电子分析天平[梅特勒-托利多仪器(上海)有限公司,感量:0.01 mg]。
1.2试药 羧甲司坦标准物质(上海脉铂医药科技有限公司,批号:12202865222),磷酸二氢钠(国药集团化学试剂有限公,批号:20200616),磷酸氢二钠(国药集团化学试剂有限公,批号:10000318),茚三酮(湖北健楚生物医药有限公司,批号:301302012),甲醇(Merck KGaA,批号:20210818),羧甲司坦微囊(自制)。
2 方法与结果
2.1溶液的配制
2.1.1pH 6.6磷酸盐缓冲溶液配制 分别精密称定磷酸氢二钠2.70 g、氯化钠1.70 g、磷酸二氢钠1.74 g,加适量水溶解后,稀释至400 mL。
2.1.2紫外分光光度法对照品溶液的制备 精密称定羧甲司坦对照品0.1 g溶于20 mL pH 6.6磷酸盐缓冲溶液中,摇匀。精密量取4 mL于50 mL容量瓶中,加入pH 6.6磷酸盐缓冲溶液稀释至刻度。
2.1.3高效液相色谱法对照品溶液的制备 精密称定羧甲司坦对照品0.1 g于100 mL容量瓶中,加入pH 6.6磷酸盐缓冲溶液水浴加热溶解并稀释至刻度。
2.1.4高效液相色谱法供试品溶液的制备 精密称定羧甲司坦微囊0.1 g溶于100 mL pH 6.6磷酸盐缓冲溶液中,过滤;精密量取滤液0.3 mL于25 mL容量瓶中,加pH 6.6磷酸盐缓冲溶液稀释至刻度。
2.1.5空白样品溶液 按处方比例精密称定各辅料适量,混匀,放入50 mL容量瓶中,加入pH 6.6磷酸盐缓冲水浴加热液溶解并稀释至刻度。
2.1.6茚三酮溶液配制 精密称定茚三酮1.0 g放入50 mL量瓶,加适量乙醇溶解,加水稀释至刻度,即得2%的茚三酮溶液。
2.2紫外-可见分光光度法的建立
2.2.1专属性的考察 精密量取“2.1.2”项下对照品溶液2 mL至25 mL容量瓶中,加入2%茚三酮溶液1 mL,加入pH 6.6磷酸盐缓冲溶液定容,摇匀;水浴加热20 min,置于冰水浴中迅速冷却。精密量取“2.1.5”项下空白样品溶液2 mL至25 mL容量瓶中,加入pH 6.6磷酸盐缓冲溶液定容,摇匀。以溶剂为空白对照,以紫外分光光度法在200~800 nm之间进行全波长扫描,比较羧甲司坦与辅料的最大吸收峰重叠程度。由图1可知,羧甲司坦与茚三酮络合物最大吸收峰在567 nm波长处,辅料在此处几乎无吸收,辅料对测定无干扰,因此选择567 nm作为测定波长。
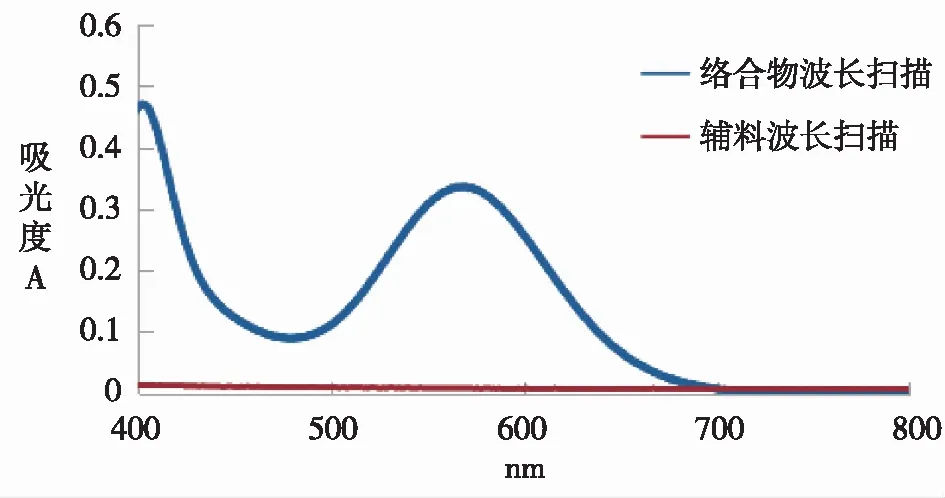
图1 羧甲司坦对照品及辅料UV扫描图
2.2.2显色时间的考察 精密称定羧甲司坦0.1 g溶于20 mL pH 6.6磷酸盐缓冲溶液中,摇匀。精密量取4 mL三份于25 mL容量瓶中,各加入2%茚三酮试剂1 mL,用pH 6.6磷酸盐缓冲溶液定容至刻度,摇匀。水浴加热15 min、20 min、25 min及30 min,置于冰水浴中迅速冷却,观察颜色变化。分别对羧甲司坦对照品溶液加热15 min、20 min、25 min及30 min后,发现加热15 min时溶液变为紫红色,冷却至室温变为无色透明,显色反应进行不完全,未测出吸光度值;加热20 min、25 min与30 min时溶液变为紫红色,冷却至室温溶液不褪色,显色反应进行完全且测定结果稳定,因此以20 min作为显色时间。
2.2.3显色剂的用量考察 精密称定羧甲司坦0.1 g溶于20 mL pH 6.6磷酸盐缓冲溶液,摇匀。取4 mL三份溶液于25 mL容量瓶中,分别加入2%茚三酮试剂0.5 mL、1.0 mL及2.0 mL,加入pH 6.6的磷酸盐缓冲液定容至刻度,摇匀。水浴加热20 min,置于冰水浴中迅速冷却,以溶剂为空白对照,在最大吸收波长处测定吸光度值,以吸光度值恰当者确定显色剂用量。由表1可知,随着显色剂的用量的增加,吸光度值呈上升趋势,以1 mL显色剂用量时吸光度值最佳,因此选择1 mL作为显色剂用量。

表1 显色剂用量测定结果
2.2.4标准曲线的制备 分别精密量取对照品溶液2.0 mL、2.5 mL、3.0 mL、3.5 mL、4.0 mL、4.5 mL及5.0 mL于25 mL容量瓶中,各加入1 mL 2%茚三酮溶液,以pH 6.6磷酸盐缓冲溶液定容,摇匀。水浴加热20 min,迅速置于冰水浴中放置至室温,以溶剂为空白对照,在最大吸收波长处测定吸光度,以吸光度值A为纵坐标,羧甲司坦的浓度C为横坐标,绘制标准曲线,得回归方程:A=0.0224c-0.5804,R2=0.999,在32.10~80.24 μg/mL浓度范围内,羧甲司坦的浓度与吸光度呈良好的线性关系。
2.2.5精密度的测定 分别精密量取2.0 mL、3.0 mL及4.0 mL的对照品溶液,加入1 mL 2%茚三酮溶液,用pH 6.6磷酸盐缓冲溶液定容,摇匀,水浴加热20 min,迅速置于冰水浴中放置至室温。以溶剂为空白对照,在最大吸收波长处测定吸光度值,1天内测定5次,计算日内精密度;每天测一次,连续测定3次,计算日间精密度,计算RSD。精密度测定结果:日内精密度的吸光度RSD分别为1.52%、1.36%和1.13%;日间精密度的吸光度RSD分别为4.65%,5.85%及6.85%,结果表明精密度良好。
2.2.6重复性的测定 精密量取对照品溶液6份,各3.0 mL至25 mL容量瓶中,加入1 mL 2%茚三酮溶液,加入适量的pH 6.6磷酸盐缓冲溶液,定容,摇匀。水浴加热20 min,迅速置于冰水浴中放置至室温,以溶剂为空白对照,在最大吸收波长处测定吸光值,计算RSD。重复性测定结果如表2所示:测定结果的RSD为3.2%,结果表明重复性良好。

表2 重复性实验结果
2.2.7稳定性的测定 精密量取对照品溶液3.0 mL至25 mL容量瓶中,加入1 mL 2%茚三酮溶液,加入pH 6.6磷酸盐缓冲溶液,定容,摇匀。水浴加热20 min,迅速置于冰水浴中放置至室温,以溶剂为空白对照,在0、2、4、6和8 h测定吸光值,计算RSD。稳定性测定结果如表3所示:测定结果的RSD为3.31%,结果表明样品稳定性良好。
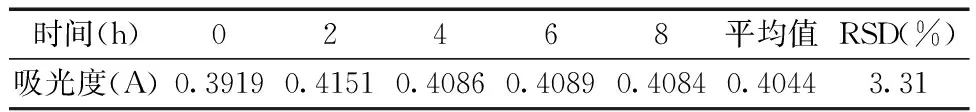
表3 稳定性实验结果
2.2.8回收率的测定 精密量取羧甲司坦对照品溶液2.0 mL、3.0 mL和4.0 mL于三个25 mL容量瓶中,各加入适量空白样品溶液,加入1 mL 2%茚三酮溶液,用pH 6.6磷酸盐缓冲溶液定容,摇匀,水浴加热20 min,迅速置于冰水浴中放置至室温,以溶剂为空白对照,在最大吸收波长处测定吸光度值,计算RSD。由表4结果显示:羧甲司坦的平均回收率为96.65%,RSD%为1.48%,表明其回收率良好。
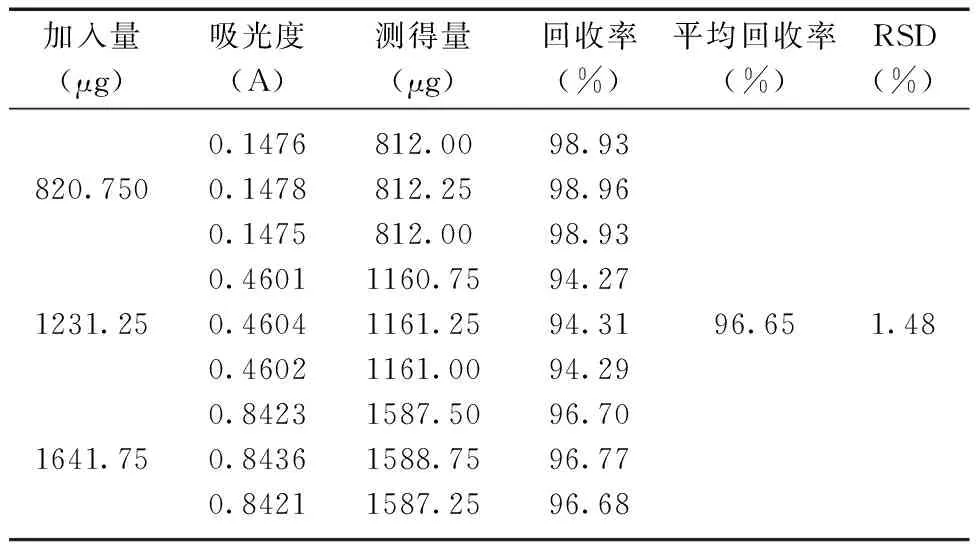
表4 回收率实验结果
2.2.9样品含量测定 分别精密称定三批羧甲司坦微囊各0.1 g溶于20 mL pH 6.6磷酸盐缓冲溶液中,过滤。分别精密量取滤液0.5 mL置于25 mL容量瓶中,各加入1 mL 2%茚三酮溶液,加入pH 6.6磷酸盐缓冲液,定容,摇匀。水浴加热20 min,迅速置于冰水浴中放置至室温,以溶剂为空白对照,在最大吸收波长下测定吸光度值。利用回归方程计算各样品的羧甲司坦含量。自制羧甲司坦微囊的含量测定结果如表5所示:结果表明羧甲司坦缓释微囊的含量测定结果符合要求,建立的含量测定方法可靠。
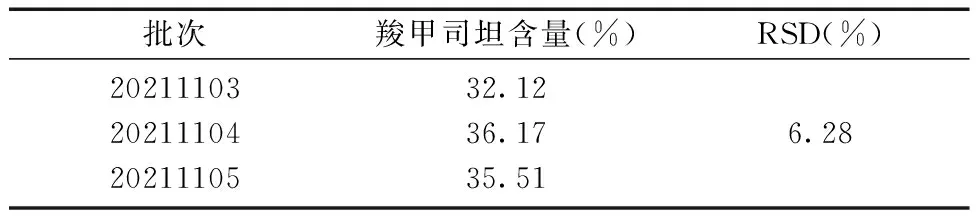
表5 羧甲司坦缓释微囊含量的测定结果
2.3高效液相色谱法的建立
2.3.1色谱条件 色谱柱:WondaCract ODS-2 C18(250 mm × 4.6 mm,5 μm);流动相:pH 6.6磷酸盐缓冲溶液;流速:1 mL/min;检测波长:210 nm;柱温:35 ℃;进样量:20 μL。
2.3.2专属性 精密量取“2.1.3”“2.1.4”“2.1.5”项下对照品溶液,供试品溶液,空白样品溶液,各进样20 μL,记录色谱图。由图2、图3和图4可知,在210 nm下扫描,羧甲司坦微囊供试品和羧甲司坦对照品保留时间为2.93 min左右,辅料对其测定无影响,专属性良好。
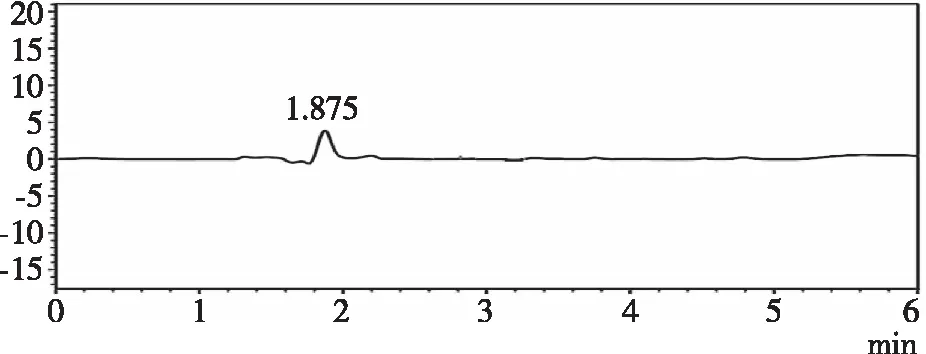
图2 空白样品溶液色谱图
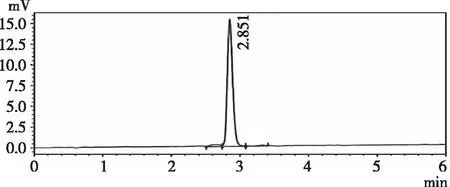
图3 供试品溶液色谱图
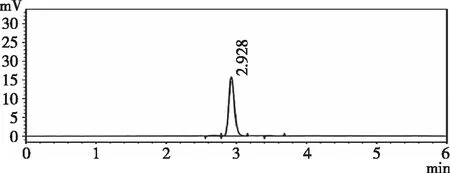
图4 对照品溶液色谱图
2.3.3标准曲线的制备 分别精密量取羧甲司坦对照品溶液0.1 mL、0.2 mL、0.3 mL、0.4 mL、0.5 mL,0.6 mL和0.7 mL置于25 mL容量瓶中,用pH 6.6磷酸盐缓冲溶液稀释至刻度,摇匀。注入高效液相色谱仪20 μL进样,在210 nm检测波长下,测定峰面积。以峰面积值A为纵坐标,羧甲司坦的浓度C为横坐标,绘制标准曲线,得回归方程:A=7432.5c-8452.1,R2=0.9994,在4.08~28.56 μg/mL浓度范围内,羧甲司坦的峰面积与浓度呈良好的线性关系。
2.3.4精密度的测定 分别精密量取羧甲司坦对照品溶液0.2 mL、0.4 mL和0.6 mL置于25 mL容量瓶中,用pH 6.6磷酸盐缓冲溶液稀释至刻度,摇匀。注入高效液相色谱仪20 μL进样,测定峰面积,一天内测5次,计算日间精密度;每天测一次,连续测定3次,计算日间精密度,计算RSD。精密度实验结果如表6和表7所示:日内精密度的峰面积RSD分别为0.59%、0.5%和0.6%,日间精密度的峰面积RSD分别为0.61%、0.37%和0.29%,结果表明精密度良好。
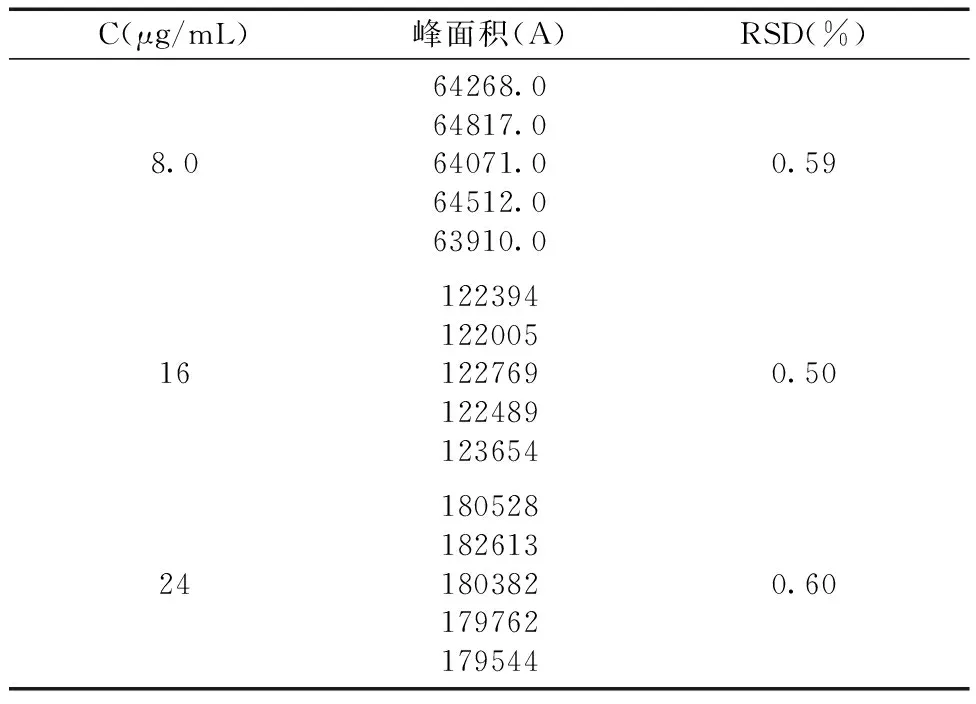
表6 日内精密度实验结果
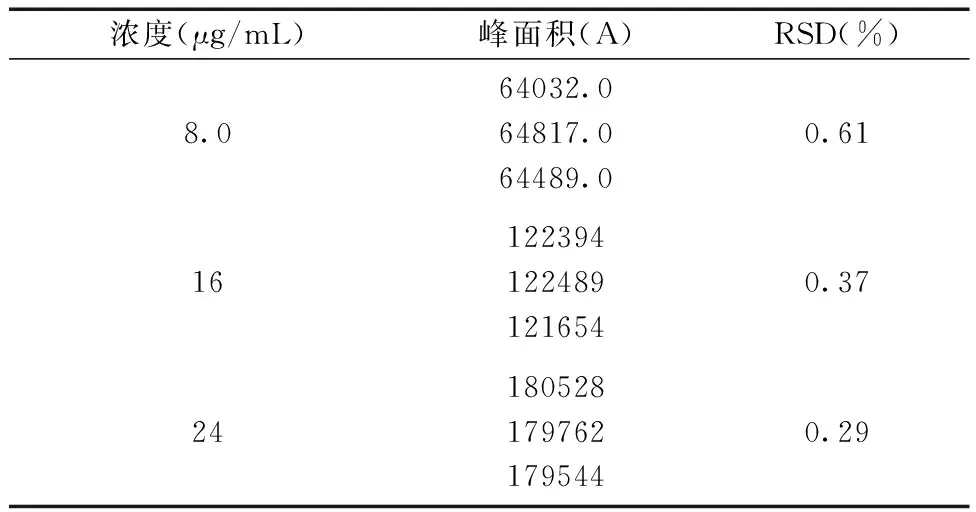
表7 日间精密度实验结果
2.3.5稳定性的测定 精密量取羧甲司坦对照品溶液0.5 mL,分别于0、2、4,6、8、12 ,24 h内注入高效液相色谱仪20 μL进样,测定峰面积,计算RSD。稳定性测定结果如表8所示:峰面积测定结果的RSD为1.02%,结果表明样品稳定性良好。
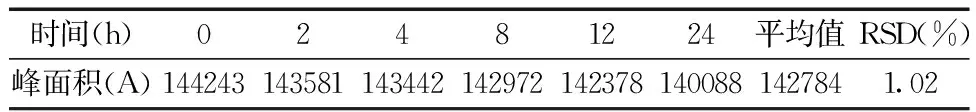
表8 稳定性实验结果
2.3.6重复性的测定 分别精密量取羧甲司坦对照品溶液0.5 mL 6份,分别注入高效液相色谱仪20 μL进样,测定峰面积,计算RSD。重复性测定结果如表9所示:峰面积测定结果RSD为0.56%,结果表明重复性良好。
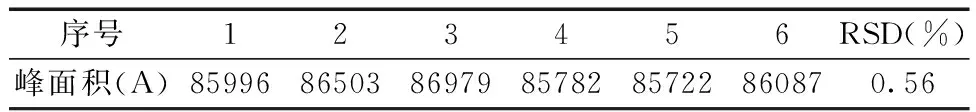
表9 重复性实验结果
2.3.7加样回收率的测定 精密称定已知含量的羧甲司坦微囊共9份,向其中分别加入相当于羧甲司坦微囊含量 80%、100%和120%的羧甲司坦对照品溶液,各3份平行样。分别注入高效液相色谱仪20 μL测定,计算回收率,计算RSD。回收率测定结果如表10所示:羧甲司坦的回收率为95.01%,RSD为1.90%结果表明回收率良好。
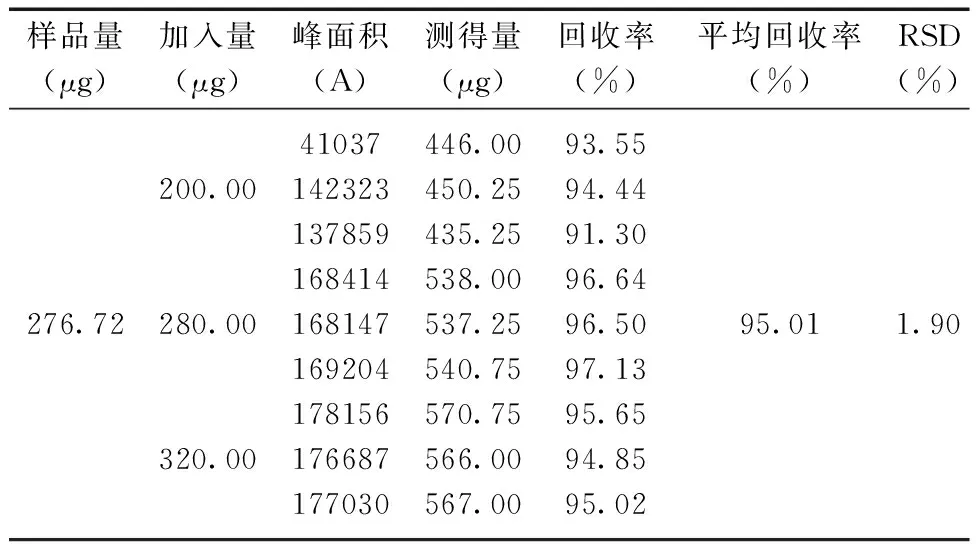
表10 回收率实验结果
2.3.8样品的含量测定 精密称定3批自制羧甲司坦微囊,按照“2.1.4”项下供试品溶液制备方法制备样品溶液3份,分别注入高效液相色谱仪20 μL,测定峰面积,根据回归方程计算羧甲司坦微囊的含量。计算RSD。自制羧甲司坦微囊的含量测定结果如表11所示:结果表明羧甲司坦缓释微囊的含量测定结果符合要求,建立的含量测定方法可靠。
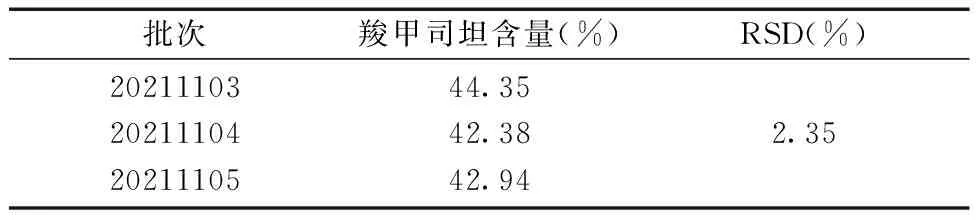
表11 羧甲司坦微囊含量测定的实验结果
3 讨 论
将药物制成缓释制剂不仅可维持血药浓度的稳定,提高患者依从性以及生物利用度,同时也可减少给药总量[7]。本研究通过建立羧甲司坦微囊含量测定发法,以为后期羧甲司坦微囊处方、制备工艺的优化提供了实验依据。既往研究[8-9]表明,应用HPLC测定羧甲司坦含量时可予pH 2.0磷酸盐缓冲液和pH 3.0磷酸盐缓冲溶液作为流动相,但本研究考虑流动相pH过低易导致色谱柱耐受性降低,极大降低其使用寿命。尽管羧甲司坦可溶于热水,但易导致其峰面积值极不稳定。本研究前期实验表明羧甲司坦在pH 6.6磷酸盐缓冲液稳定性良好,且羧甲司坦主峰和杂质峰分离较好,溶剂和辅料对主峰无影响。故最终采用pH 6.6磷酸盐缓冲溶液作为流动相。从表5及表11的结果表明,本研究所采用的两种含量测定方法得出的含量测定结果均符合要求,建立的含量测定方法可靠,因此紫外分光光度法和高效液相色谱法均可作为羧甲司坦微囊含量的测定方法。虽然高效液相色谱法的RSD较紫外分光光度法小,精密度、灵敏度较UV更高,但UV相对于HPLC具有操作简便,仪器简单,分析速度快,成本低等优点,因此,应充分利用两种方法。
综上所述,本试验建立了羧甲司坦微囊的紫外分光光度法和高效液相色谱法两种含量测定方法,其操作方法简单,方便,稳定,可靠,重现性好,为该制剂的质量控制提供了实验依据。
