“可折叠性”在酶智能设计改造中的应用研究
——以AlphaFold2为例
2023-07-10孟巧珍郭菲
孟巧珍,郭菲
(1 天津大学智能与计算学部 计算机学院,天津 300350; 2 中南大学计算机学院,湖南 长沙 410000)

酶一般是功能性的蛋白质,在各种生物反应中作为生物催化剂参与,是生物细胞发挥功能不可或缺的部分。经过漫长的岁月进化,天然酶为了适应自然环境而拥有了特定的功能[1-2],一般适宜在温和环境下且具有特定作用。由于具备高效特定作用,且无污染的特性,酶非常受工业生产研究人员的青睐。例如用于酿酒的酵母菌、用于降解塑料的酶等等,都是酶分子应用在工业领域中的经典例子。但实际工业生产过程中,发现在工业环境中直接应用天然酶并没有达到满意的效果。错误的折叠、出现副产物、功能不适宜等缺陷对酶在工业行业的应用发出了挑战[3]。
要想解决这一问题,必须对酶进行改造或者设计新酶来满足特定的工业环境或者功能需求。那么,认识酶的结构与功能的关系是非常重要的[4]。传统的酶改造过程涉及到修改酶的基因,使其在细胞中被成功表达纯化[5]。然后对得到的突变体进行试验验证是否能提高性能。这期间的时间、人力成本是巨大的,而且成功率非常低。随着人工智能技术的发展,利用计算方法辅助指导酶的改造或者设计开始成为主流[6-10]。计算算法的快速实现,极大地降低了遍历穷举整个可能计算空间的搜索,同时利用优化算法很容易寻找到可行解。例如中科院微生物研究所吴边课题组[11]使用多种计算工具,根据塑料降解酶的序列从保守性、结构能量值等角度筛选可能存在的突变位点,获得了塑料降解酶PETase的突变体DuraPETase。该突变体的熔融温度提高了35 ℃,温和温度下对塑料降解酶的降解能力提升了23%。根据特定的改造或者设计目标,智能计算方法一般是基于酶的序列或者结构挖掘和酶功能之间的映射关系,并希望借此能了解酶的各种作用机制,比如催化作用、特异性结合能力等。
那么,对于设计或者改造后的新酶,是否可以按照实验要求折叠成给定的构象,实现要设计的功能?这个能力一般称为“可折叠性”[12-13]。实验验证是将新酶序列在大肠杆菌中纯化表达,同时测定是否具有给定的功能。但是,现在很多工作随机生成新酶,可以产生大量要求的序列。这些序列如果都通过实验室测定其是否合理,并不符合通过计算手段降低实验成本的初衷。迄今为止,尽管从头酶设计有了诸多成果,但大多都表现出低效率。有研究表明错误折叠是大多数酶设计工作失败的原因。如果在设计或者改造的过程中,考虑加入结构约束的话,则在很大程度上可以提高新酶的“可折叠性”。最近5年来,基于人工智能与数据驱动技术的蛋白质结构预测取得了一系列的突破性进展[14-16]。例如,AlphaFold2[14]预测了人类蛋白组的98.5%蛋白结构,极大丰富了蛋白结构数据并促进对人类生命机制的研究。实际上,蛋白质结构预测实际上可以被称为“逆式”的蛋白质设计。那么,在蛋白质设计领域,蛋白质结构预测这些相对成熟化的工具,是否能从结构约束角度促进酶的改造设计工具更快速且精确化促进酶的“可折叠性”研究呢?
本文聚焦于智能算法改造设计新酶这一应用背景,首先对现有的研究工作从骨架设计、序列设计两个角度进行了系统性的梳理。然后介绍了成熟化的蛋白质结构预测工具的四种方法框架,并以AlphaFold2为重点介绍了相应的工作流程。蛋白质结构问题可以理解为寻找一个合适的拟合函数f,能够将序列空间映射到结构空间。因此这部分内容从四个角度来展开:①基于物理化学规则打分;②基于统计知识打分;③基于深度学习预测打分;④端到端一步式。蛋白质改造设计则分别从序列和结构两个角度挖掘和功能之间的模式(见摘要图)。最后本文总结出三种蛋白质结构预测工具在酶设计/改造中的应用场景,展示如何利用“可折叠性”帮助改造或者设计稳定且具有给定功能的酶。希望本篇文章能对如何利用正确折叠进行合理酶设计改造有所帮助。
1 酶的智能改造设计策略
人工智能在酶的设计改造过程中的应用,有助于对酶序列、功能以及结构空间的快速探索。对于酶的计算机智能辅助设计,通常集中于酶的热稳定性、耐酸碱性、催化活性、底物特异性以及酶的从头设计等方面[2]。前面几种的设计着重于对酶的功能空间的探索,提高酶的某种已有功能特性,且不影响其原有的其他功能特性。而酶的从头设计则侧重于设计一种新酶,其目标功能可能只是具有8个β“片段桶”(barrel)这样的形状要求,或者是这个“桶”从结构上更为松散的功能性要求,又或者是β片段的排列方式这种结构上的要求。这意味着酶的设计要从结构和功能上达到统一。
利用人工智能解决问题是根据已有的数据挖掘内部隐藏的看不见的模式,即序列、结构与功能之间的内在的关系映射。第一步则需要合理地将酶的描述特征提取到并表示成机器识别的模式,一般分为以下几类:基于序列的,基于结构的,基于嵌入的。基于序列的,包含一些常见的onehot编码、物理化学特性编码(疏水性、电荷等)、进化保守性、AA-index[17]、zScales[18]等。基于结构的,包含一些基于统计的残基对间的接触势、相邻结构域的类型及物理化学性质、骨架扭转角度、键长、距离活性位点的远近等[19]。而基于嵌入的,是指模型通过在大量蛋白质家族序列或者结构上进行类似于“完形填空”的训练过程中,学习到序列/结构邻居的上下文信息。在此过程中,模型学习氨基酸的有意义的中间表示,并提炼出每个氨基酸位置周围的重要结构环境,比如ProtVec[20]、ESM-1V[21]、TAPE[22]、dMaSIF[23]等。接下来需要构建合适的模型预测或者生成目标。这部分的差异,可参考文献[24]。接下来根据目标从酶的智能改造和设计两部分展开。
1.1 酶的智能改造
酶的智能改造通常指的是在对酶的催化机制、空间结构、物化属性等有一定了解的基础上,利用计算手段有目的地对酶的功能进行改造。对于任意的一条酶序列,可能的突变方案都是非常庞大的,且无法在实验室逐一验证所有可能的突变方案是否合理有效。采用人工智能技术寻找酶的可能突变位点以及对突变位点组合,能够快速地实现高通量筛选,减少生物化学实验成本。这里仅结合人工智能探讨现有对酶的功能改造相关工作。
利用酶的序列以及功能性指标数据对,构建模型,然后利用模型指导酶分子改造。其构建的模型输入一般是基于序列或者结构提取的描述符,输出则是蛋白质适应性的预测目标,一般对应于要改造的具体功能性指标。一旦模型建立,即可通过预测大量突变序列的性能快速筛选不理想的突变体。以Frances H.Arnold团队[25]发表在PNAS上的工作为例。该工作主要是改造一氧化氮双加氧酶(NOD)立体选择性,并选择多个机器学习模型去构建NOD的立体选择性催化模型,包括但不仅限于K最近邻、线性模型、决策树、随机森林,将76%(S)-ee初始突变体提升至93%(S)-ee及反转至79%(S)-ee。中科院微生物研究所吴边团队[11]提出一种新型蛋白质稳定性计算设计策略GRAPE。该策略对传统筛选突变体策略进行补充,并通过系统聚类分析对得到的单点有益突变进行聚类,同时结合贪婪算法进行网络迭代叠加,大幅度规避了以往遇到的累积突变所带来的负协同相互作用。设计出的突变体DuraPETase可在中等温度下有效降解塑料,为酶的设计的计算策略提供了非常重要的方向。当特定类型的酶数据比较小的时候,可以借助在大量通用酶类数据上的预训练模型来学习氨基酸对之间的相互作用关系或者邻居结构环境信息,指导后续的酶改造任务。这种方法的好处是可以根据特定任务在具体的数据集上对预训练模型进行微调,以适应于不同的小数据集的下游任务。2021年提出的Low-N模型充分利用了UniRep中大量的蛋白质序列,通过无监督语言预训练任务提取了蛋白质的一般功能特征,然后在特定家族序列上微调,进一步捕捉到了该家族的特异性特征[26]。通过上述方式得到的蛋白质表示,仅需要少量的序列和目标功能的数据,就可以训练一个简单且有效的监督模型。将该模型应用到实际中,最少仅需24个avGFP突变体的数据集,就设计出了新的荧光蛋白,可以与高保真且高通量的蛋白质工程产物sfGFP相媲美。Low-N以较少的数量实现了蛋白质序列到功能模式的转变。类似工作还有文献[27]中提到的SEMA。
除此之外,随着日益丰富的结构数据与逐渐成熟的深度网络学习能力,从酶的结构数据集中直接挖掘结构与功能之间的关系也成为可能。2022年,得克萨斯大学奥斯汀分校McKetta化学工程系教授Hal S.Alper[28]结合人工智能技术和酶工程,改造出一系列塑料降解酶的变体,相关工作发表在Nature上。其中最优秀的突变体FAST-PETase优于现有的PET降解酶的变体的降解效率,且能在更广泛环境中具有较好的活性,证明了在工业规模上酶塑料回收的可行途径。该方法首先筛选有效突变位点的方法是利用一个深度学习算法MutCompute[19]来有效过滤筛选突变位点 。MutCompute通过一个3D的自监督的卷积网络模型,对每一个残基构造一个局部微环境,统计该环境中原子(C、H、O、N、S)出现的次数、电荷、溶剂可达面积来编码该局部环境,最后预测每个残基的序列类型(分类问题)。根据该残基一个已有突变体上的预测概率值与在野生型中的概率差异值大小,衡量出残基在野生型结构中的“不匹配度”(disfavoured),进而筛选出这种得分较大的突变位点,结合以往文献中报道的有效突变位点以及活性口袋位点,指导后续进一步筛选有效组合突变。该方法捕获了由结构决定的功能模式的指导转化,筛选条件是该残基在给定的蛋白质折叠环境中适配的能力。相比单纯使用序列的模型,考虑残基在结构环境中是否适配或从已有结构数据中挖掘这种规律,约束了改造酶的合理性并且增加了可能的改造位点方案。类似的工作还被应用在TEM-1 β-内酰胺酶和白色念珠菌磷化异构酶(CaPMI)中[29]。
实际上在酶改造过程中,序列和结构信息并不是互相割裂的。Connor W.Coley组提出一种将结构约束在序列表示上,就是一种有效的思路。相比仅用ESM-1b[30]提取蛋白质序列的平均池化模式得到的序列特征,融入离酶活中心远近的结构性差异构建的池化策略,则在增强酶的嵌入性表达的同时还提高了酶活性预测任务的模型性能[31]。丰富的酶结构信息,是非常重要且有效的(参见上面加入结构约束之后几个工作的性能提升)。随着AlphaFold2等高精度有效的蛋白质结构预测方法的提出,如何结合预测出来的海量结构数据扩展对酶的功能改造,是具有研究价值的。
1.2 酶的智能设计
酶的从头设计是指创造出自然界中不存在,具有新的功能、结构或者形状的酶。在人工智能技术没有被引入到这个领域之前,大多数酶的设计是构建基于物理或者统计的模型去拟合力场(这一部分的基本思路和蛋白质折叠一致)。本小节根据不同的设计目标以及任务需求,从主链结构设计、氨基酸序列设计两部分展开,着重探讨智能计算算法给蛋白质设计领域带来的新思路(如表1)。
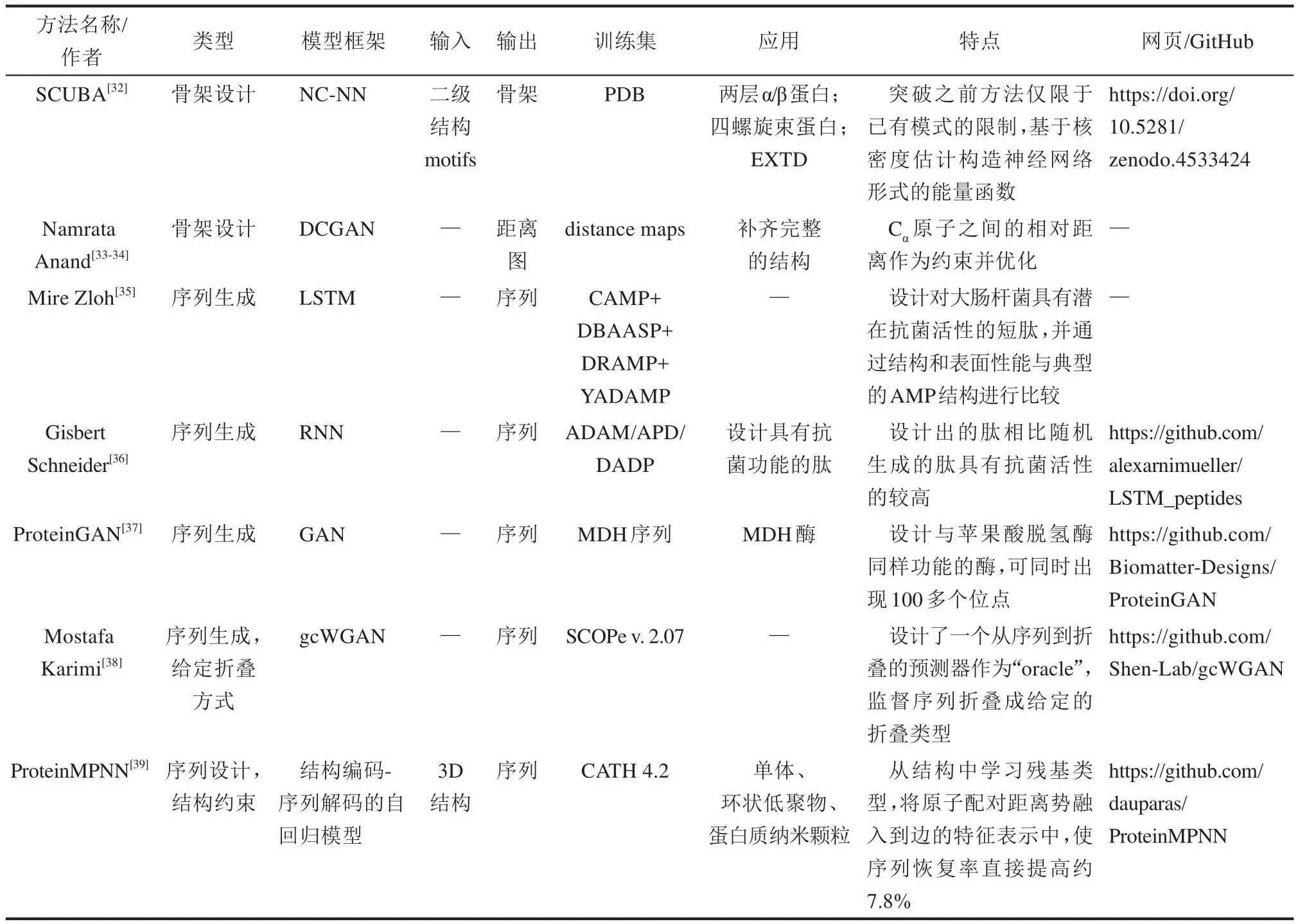
表1 蛋白质设计工具汇总Table 1 Summary of protein design tools
1.2.1 主链结构设计
主链结构设计,指的是设计出符合预先定义的结构拓扑约束(例如:二级结构基本单元的组成以及顺序、相对位置等)。这里介绍一个非常典型且有突破性的工作,SCUBA[32]。该工作由中国科学技术大学刘海燕和陈泉团队提出,是一个具有高自主可设计性的主链设计算法,且并不依赖侧链类型。该算法在结构数据中基于核密度估计构造神经网络形式的能量函数来捕获高阶相关关系,可在不确定序列(即设计的能量函数不依赖于侧链,充分考虑柔性)的情况下,连续广泛搜索主链结构空间,突破之前方法仅限于已有模式的限制。再辅以该团队提出的给定主链设计序列的能量统计模型ABACUS[48],形成了一套全新的蛋白质自主设计新路线。
此外,Namrata Anand陆续提出基于生成对抗网络(generative adversarial network, GAN)[49]实现蛋白质骨架设计的工作,从生成模型的角度考虑蛋白的骨架设计。发表在2018年的NeurIPS[33],利用DCGAN(deep convolutional GANs)[50]模型生成Cα原子之间的相对距离图(考虑到平移旋转不变性),将该配对距离约束引入到折叠成给定结构的可微问题中,并采用交替方向乘子法(alternating direction method of multipliers, ADMM)优化该凸规划问题[33]。紧接着2019年发表的另一个工作也采用GAN实现给定距离约束下骨架设计,只是后面的精细化调整有所不同[34]。
1.2.2 氨基酸序列设计
氨基酸序列设计,则是在蛋白质结构已知的情况下,设计其相应的侧链类型,也就是氨基酸序列。根据在设计过程中给出的约束不同,可以采用不同的方法来设计序列。
当从功能上约束设计的序列时,可以采用序列生成方法,在具有给定功能的酶序列数据上挖掘残基间的模式直接生成新酶的序列。常用的生成模型有长短期记忆网络(long short-term memory,LSTM)[51]、GAN、变分自动编码器(variational autoencoder,VAE)[52]、Transformer[53]等。Mire Zloh课题组[35]构建了基于LSTM的生成模型和双向LSTM分类模型,设计了对大肠杆菌具有潜在抗菌活性的新型的抗菌短肽序列,经过分类模型的预测发现设计出的肽序列被认为具有抗菌功能的概率在70.6%~91.7%,且其三维构象表现出具有两亲性表面的α-螺旋结构[35]。Gisbert Schneider课题组[36]同样使用LSTM从螺旋抗菌肽序列上捕获数据的模式并将学习到的上下文信息运用于抗菌肽序列的生成。Aleksej Zelezniak课题组[37]提出ProteinGAN,利用GAN学习到大量天然蛋白质序列的多样性并进而生成具有特定功能的酶序列。以苹果酸脱氢酶(MDH)为例,作者在该酶家族序列上进行训练并设计出具有相同功能酶的序列,其中有突变位点超过100个的设计序列,其活性与天然酶的活性相近。
同样,可以采用结构约束来指导进而设计氨基酸序列。这种情况下,设计的氨基酸序列能否折叠成目标的蛋白质结构是至关重要的指标。最近被称为新一代Rosetta蛋白设计内核的Rosetta MPNN “Mover”,突破了传统的Rosetta设计范式“inside-out”模式。该方法ProteinMPNN由David Baker组提出,基于structured-Transformer[54],采用了结构编码-序列解码的自回归模型框架,将原子配对距离势融入到边的特征表示中,使序列恢复率提高约7.8%[39]。ProteinMPNN对根据幻想的主链进行蛋白设计,其中96条蛋白质序列在大肠杆菌体系中可以被大量可溶表达,且成功结晶一个与设计结构高度一致的设计蛋白。同时,ProteinMPNN对单体、同源二聚体、异二聚体结构进行设计,其序列恢复率均在50%以上,其中核心区域的恢复率高达90%~95%。中国科学技术大学刘海燕和陈泉团队[40]提出的ABACUS-R完全基于深度学习算法实现给定骨架设计氨基酸序列,不再依赖于传统能量项构建,并且序列恢复率高于ABACUS计算的,在测试集上基本可以达到50%[40]。其主要思路是在给定骨架的情况下,通过编码-解码(encoder-decoder)框架学习在给定残基的结构特征以及周边结构环境的特性预测该残基的序列类型(侧链)。值得一提的是,ABACUS-R采用多任务学习,不仅仅学习该残基的类型,还同时预测其二级结构、溶剂可达面积、B-factor以及一些结构构象扭转角任务。这些辅助任务的设计不仅提高了模型设计序列的能力,还隐式地在序列设计中加入了实时的结构约束。实验验证设计了3个天然骨架的蛋白序列设计并做了相应的实验验证。最后通过ABACUS-R设计出了可以成功表达且折叠成相应的三维结构的蛋白质序列,充分证明了绕过建模侧链模型的蛋白质设计是可行的。卜东波课题组[47]提出ProDesign-LE也是基于Transformer框架,通过计算序列类型是否符合给定的局部结构环境来设计蛋白序列。在实验中为CATⅢ酶设计的5条序列中,有3条可以成功表达且可溶。许锦波课题组[55]提出的一种基于骨架设计蛋白序列的方法,基于生成SE(3)等变模型,显著改进了现有的自回归方法。Mostafa Karimi组[38]提出gcWGAN探索生成给定折叠条件下的序列,使序列折叠成给定的方式。构造一个基于DeepSF[56]的快速从序列预测折叠模式的模型并实时反馈监督序列是否可以正确折叠,这个模型被称为“Oracle”。Po-Ssu Huang组的Namrata Anand[57]直接从蛋白质骨架结构信息中预测侧链氨基酸类型,从而学习到一个基于自回归的自动的神经网络能量来指导后续的序列设计。在实际的TIM-barrel设计中,设计出的序列中有两个成功结晶且与设计的骨架高度一致。
总的来说,对于酶的智能设计,人工智能方法的设计相比传统基于力场的模式带来更高的成功率,且更加快速(ProDesign[47]仅需30 s即可设计一条少于100长的蛋白序列)。根据不同任务需求,可以实现酶的全新骨架设计和酶序列的从头设计。同时将二者结合起来可以形成一套按需从头设计酶的流程。酶设计中直接从给定结构建模设计序列的方法(类似于MPNN),本质上是为了寻求一条序列使结构能量最低。但是给定一条序列,其所能折叠成的状态有很多,目标结构不一定是设计的序列所能折叠成的最低的能量结构。因此现今从头酶设计中最关键的是后续对新酶的折叠能力评估。设计的新酶序列在后续的实验中评估能否折叠或者折叠成给定的目标构象,这是在实际应用中最关注的问题。因此,在设计酶的过程中,利用“可折叠性”作为指标过滤设计序列,有助于设计更高质量的酶,减少了实验室对酶序列的后续验证,从而降低成本。
2 蛋白质结构预测方法
从上面的讨论中我们可以看到人工智能极大促进了酶在改造和设计方面的发展。但是对于设计或者改造后的新酶,其是否可以如期折叠成给定的结构,其实是其能否执行相应功能的关键性问题。那么,如何衡量“可折叠性”?一般是通过一系列的实验操作观察其最后是否折叠或者折叠后与目标结构的结构相似性(TMscore得分)。但是实际上,如果在设计或者改造的过程中同时考虑“可折叠性”,就会大大提高最终酶的质量。因此,成熟且高精度的蛋白质结构预测工具是极其有必要的。
蛋白质折叠问题是Science杂志指出的人类在21世纪需要解决的125个科学前沿问题之一。蛋白质分子作为细胞这所天然工厂中不可或缺的主力,根据周边环境的变化,通过展开与折叠过程的不断转移,实现结构从变性到天然状态下稳定紧凑折叠结构的变化,从而实现蛋白质序列信息的解码,发挥蛋白质的功能。蛋白质结构预测问题可简单用数学公式简单表述为:g=f(s)。其中s表示蛋白质序列,g表示蛋白质结构,求解蛋白质结构就相当于在求解函数f的表达式。函数f越精准,预测的结构越准确。显而易见,是否能找到一个“完美”的能量打分函数f,能正确表达在折叠过程中各个原子空间之间的能量变化、位置,从而正确区分天然构象和其他构象,是整个蛋白质结构预测问题中的关键。本文着重从4个角度对如何构建函数f来进行阐述:基于物理化学打分,基于统计知识打分,基于深度学习预测打分以及“一步式”构建。前三种方法均倾向于寻找完美的“能量函数”(或者称之为“打分函数”),更好地模拟原子从杂乱而无序的状态到相互作用进而形成稳定折叠状态过程中的各种力场变化。得到具有一定规律的“打分函数”后,一种是依据热力学系统中能量越低越稳定这一基本原则,随机模拟寻找具有最小能量的构象,另一种是将“打分函数”转为可微函数,将蛋白质构象预测转化为数学中的最优化问题寻找最优解(即最优构象)。那么这三种的差别则体现在构建能量函数的规则或者手段上。一般在实际情况中会适当从这三种方法中挑选合适的能量项组合,以寻找更加适合的复合折叠能量函数。最后一种则是直接实现端到端的蛋白质序列-结构模式的深度挖掘,一步式实现从蛋白质序列到结构的输出。
2.1 基于物理化学打分
基于物理的能量项,通过描述原子在折叠过程中原子内部之间相互作用以及蛋白质分子与溶剂分子之间的相互作用,来模拟构象的最终能量。一般包括成键作用和非成键作用[58]。后者主要包括氢键、范德华力、静电力等,前者则包含一些二面角、键角、键长等势能[59-61]。但是在实际过程中,由于我们对蛋白质折叠机制尚未完全理解,例如哪些相互作用力对折叠是重要的、不同相互作用力的叠加是否是有益的,这就导致在设计能量函数的时候并不一定合适。
2.2 基于统计知识打分
基于知识统计的方法,一般要求有一个大型结构数据集(类似于PDB[62]),从中统计不同原子对之间的相对位置,进而构造一个打分矩阵,得到原子对之间的打分函数。例如,在打分矩阵中,发现某种氨基酸在其相邻的0.36 nm范围内经常有一种氨基酸出现,且对方的相邻打分矩阵中也显示经常与之相邻,则能量值打分一定是较低的。从中,其实可以看出该方法要求预测的这个蛋白质结构在已有的蛋白质库中存在相似的蛋白质结构区域,即局部的某些构象出现的次数一定不低,否则这个能量项即使很高,也是有一定“偏见”的。美国密歇根大学张阳实验室[63]开发的从头预测蛋白质结构预测工具QUARK是典型的基于统计能量项的工作。QUARK分别从原子层面、残基层面、拓扑层面统计了11种基于知识的能量项,利用副本交换的蒙特卡洛搜索算法实现仅从序列出发预测蛋白质结构的工作。另一个同样由张阳实验室开发的I-TASSER,采用基于统计的能量项迭代的基于线程结构模板装配方法在近几年的Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP7-CASP15)大赛上均位列服务器组第一名[64-65]。I-TASSER采用的是基于统计的势能,包含三种类型:①通用的统计势能,特定方向(平行,反平行,垂直方向)的接触特征,手性局部结构的短程Cα原子的距离关系,相隔5个残基的局部结构特征规律等。②氢键网络。③基于线程模板的约束,包含Cα原子之间的距离约束以及侧链质心原子的接触距离约束。而与I-TASSER并驾齐驱的由美国华盛顿大学的David Baker组开发的Rosetta方法,则同时采用了基于物理能量项和基于统计的能量项,运用蒙特卡洛算法在构象空间中基于Metropolis准则随机搜索最低能量构象[66]。
2.3 基于深度学习预测打分
基于深度学习预测打分的思路其实沿袭第一种、第二种的构造思路,只是在实现过程中采用的技术手段不同。其主要手段是依赖于深度学习算法在海量结构数据中预测出不同残基组合在折叠过程中的模式(“学习”到的能量函数),从而辅助指导或者约束蛋白质的不同折叠排列方式。这里面提到的模式,在多数工作中涉及到的是残基之间的接触(contact)分布、距离(distance)分布、原子角度(orientation)分布等。而在折叠过程中,这些约束规则一旦定义,则类似于搭积木一样,很容易就可以从给定的氨基酸序列出发搭建出准确的三维结构空间。学习到基于神经网络构建的函数后,将其作为约束加入到能量项函数中,直接优化该函数并且求解最优构象或者随机寻找能量最低构象。下面通过几个典型方法的引入来理解通过深度学习预测不同的结构约束作为能量项的过程。
早期的蛋白质折叠将蛋白质三维结构中的物理接触(contact)作为约束。通过分析蛋白质序列残基的共进化信息,将序列中残基的共变关系映射到蛋白质三维空间结构中的物理接触中。共进化指的是在蛋白质家族的进化演变中,由于环境以及自身进化的需要,某些残基发生突变后,为了维持某些主要的功能或者结构不变,其他残基随之发生共同变化(co-evolution)的现象。从蛋白质家族的多序列比对(multiple sequence alignment,MSA)中统计不同位置上不同残基对共同出现的频率大小进而估计它们之间的相互作用,根据相互作用大小判断在空间结构上是否接触或排斥。主要的估计方法有:稀疏逆协方差方差估计[67],互信息最大化[68],直接耦合分析(direct coupling analysis, DCA)[69]。这样基于概率统计模型得到残基相互作用对估计量的方法,显而易见依赖于MSA的丰富程度并且难以达到满意的精度(主要是噪声以及信息的不足)。但是由于结合了全局信息,相比“孤立”预测残基对的方法,还是有了很大的突破[70-72]。随着人工智能技术的发展,解决手段就变得更为丰富多样起来,预测精度也有了突破性的进展。2016年许锦波课题组[73]提出的“RaptorX-Contact”方法首次将深度神经网络应用在蛋白质结构领域,在CASP12比赛中一举夺冠,证明了深度学习算法在该领域的可行性。该方法将残基对之间的相互作用关系看作图像问题,提取一维的序列保守性特征、结构特征以及二维的共进化特征,然后采用2D深度残差网络(ResNet)块预测残基对是否接触,协助蛋白质的从头折叠。该方法使用的ResNet网络相比前面提到的早期研究方法,捕获到了更高阶(high-order)的残基对关系,而且训练数据从单一到大量蛋白质家族上挖掘,因而精度有了明显的提升[74]。
除了上面提到的接触约束,CASP13上DeepMind提出的AlphaFold1,则将这一约束扩展到了残基间的距离约束。然后将离散化的距离预测值通过采样插值转化成可微的残基距离分布函数,进而通过直接优化该函数求解距离和角度的最优解,从而确定最终的蛋白质三维结构[15]。AlphaFold1的成功不仅仅是预测精度的显著提高,更是作为一种信号:深度神经网络可以有效识别蛋白质序列中的信号以及共进化信息的模式,并将其转化到高精度的距离分布上。考虑到三维空间的特性,trRosetta相比AlphaFold1还引入了5个角度的预测值来表示残基间的相对方向,进一步加强了残基间的约束,并且精度提高了6.5%[16]。David T.Jones组[75]提出的DMPfold,预测的是相对残基间的距离、主链氢键以及扭转角。当学习到这些约束后,类似于RaptorX,输入到crystallography and NMR system(CNS)[76]中作为约束指导蛋白质从头折叠。在2022年的CASP15上,张阳课题组在已有的I-TASSER基础上提出的D-I-TASSER算法[77],将AttentionPotential以及DeepPotential[78]两个深度学习算法预测出的高准确度的氢键(hydrogen-bond)网络、接触图以及距离图等约束加入到I-TASSER中采用的力场能量项中,然后通过蒙特卡洛模拟进行迭代的片段组装装配最终的蛋白质结构构象,该方法位列蛋白质单体单结构域比赛第一名。
2.4 端到端一步式
前面的三种本质上其实还是在拟合折叠物理力场中的各种相互作用的能量。实际上基于能量设计的方法,很难找到一个“完美的”能量函数。随着不同能量项的累积,带来的误差也随之增加。基于深度学习预测的方法中提到的“两步走”方法,虽然将复杂问题简化,但势必会带来信息的丢失。因此对于二维的表示会有更高的要求。对于这种复杂高维的相互作用,可以借助神经网络函数,直接寻找到一个更加“完美”的能量函数去拟合蛋白质分子折叠过程中的力场变化,而不是通过人工构造能量项,即直接学习到深层次的序列-结构关联关系,是近些年一些研究者的热点。随着深度学习技术的逐渐纯熟以及研究者对蛋白质结构功能的了解加深,直接基于蛋白质原始序列端到端预测蛋白质结构技术也有了质的飞跃,有力促进了研究者对蛋白机制的研究以及未知蛋白的探索。
2019年Mohammed AlQuraishi[79]提出RGN方法,首次尝试使用深度学习算法端到端从蛋白质序列直接预测最终的3D坐标,而不是通过前面介绍的“两步式”方法。其主要思想是将每个残基作为一个可微基元,然后从两个方向——N端到C端、C端到N端,预测在已有的所有残基的局部结构下当前残基加入后的空间结构,从而将整个蛋白质残基序列串联起来,得到最终蛋白质结构。这个过程中,考虑了当前残基与相邻残基之间的相互作用关系,并实现了“多个尺寸”的蛋白质表示学习。实验证明相比CASP11、CASP12上排名第一的Server组来说,该方法在对于具有新折叠的自由建模中表现优异。但是该方法输入是蛋白质序列one-hot编码以及位置保守性特异矩阵(position-specific scoring matrices,PSSM),然后通过LSTM去实现序列的编码框架,预测出每个残基的扭转角参数。PSSM相比前面提到的MSA中提取的共进化信息,并不包含残基对间的相互作用,只着重单个残基在单个位置上的进化保守性。因此,该方法:①依赖PSSM矩阵的特征准确性;②忽略残基对间的相互作用(MSA中共进化信息不是线性的,成本高,且不适合RGN的循环方法)。而之后在CASP14比赛上,DeepMind提出AlphaFold2[14],完全抛弃了AlphaFold1传统的“两步式”思路,通过图推理的方式直接实现了“端到端”(end-to-end)的蛋白质结构预测方法,转变了结合人工智能研究蛋白质结构研究新范式。因此,由该方法引发的“AI蛋白质折叠”被MIT Technology Review评为“全球十大突破性技术”。AlphaFold2主要由神经网络EvoFormer和结构模块两部分组成。EvoFormer中序列信息和从MSA中抽取的进化特征之间进行信息交换,直接推理出在空间和进化关系中残基对的配对表征。结构模块则用于将得到的特征转化为三维坐标结构。AlphaFold2的优势在于信息流之间的注意力机制,包括从MSA中学习到配对特征表示与序列上每个残基的特征表示之间的相互信息交流(基于注意力机制),通过几何空间约束形成的具有共残基的相互作用残基对之间的信息交流(三角注意力机制)。得到更新后的配对残基特征以及单残基特征后,通过结构模块不断迭代更新坐标系预测当前残基和相邻残基之间肽键的角度和距离偏移,最终得到整个蛋白质的全局笛卡尔系坐标。平均自由建模精度(GDT打分)达到80以上,而在CASAP13(AlphaFold出现)之前,这个值最高是40左右。
对于AlphaFold2来说,尽管其预测精度在CASP14上表现惊人,但是后续研究者陆续发现其高度依赖共进化信息以及模板信息,而且对于一条蛋白质在CPU上进行搜索需要大概30 min[80]。因此,从2022年起,陆续有工作直接从已有序列出发,不再显式利用共进化信息,通过大规模语言预训练任务(一般采用的模型框架是Transformer)在海量蛋白质序列数据库中学习残基的表示以及残基对的表示关系,直接输入到AlphaFold2的结构模块中,输出蛋白质结构的3D坐标[80-83]。这些方法相比基于共进化的方法(AlphaFold2)来说最显著的优势是速度上提升了一个数量级,对于宏基因数量组的蛋白质结构从时间尺度上成为可能。Meta-FAIR提出的ESMFold[80],不仅推理速度比AlphaFold2快,同时对于低复杂度序列的推理精度与AlphaFold2相当。除此之外,还有Ratul Chowdhury提出的RGN2[83],华深智药提出的OmegaFold[82],上海天壤科技开发的TRFold方法,山东大学杨建益团队提出的trRosettaX-Single[81]等方法。上述方法基本思路差别不大,各个团队在模型框架上存在一些技巧的差别。例如,trRosettaXSingle采用了知识蒸馏的思想,利用基于进化的模型作为“老师”去指导仅基于序列的“学生”模型获得一个比较理想的结果。这些方法预测一个蛋白根据计算资源和长度的不同,计算时间基本在毫秒到秒级,同时不依赖于共进化信息。这种优势对于缺少同源信息的酶设计改造来说,是非常有必要的。
通过上面的介绍可以发现,现有蛋白质折叠预测问题借助人工智能技术,已经取得了突破性的进展。直接基于蛋白质序列高精度预测蛋白质结构已经成为可能。那么,如何借助这股“东风”助力酶智能设计改造,则是结构到功能这一新研究范式的主要研究问题。同时,我们认为关注设计或者改造的新酶是否具有“可折叠性”,是在考虑实际改造设计酶在合成落地过程中的关键性问题。
3 蛋白质折叠在酶智能设计改造中的应用
第一部分中提到,对于酶的改造和设计这两个应用场景,设计新酶的折叠能力是至关重要的。不论是在给定结构还是在给定功能约束下,设计的新酶如果不能正常折叠或者折叠后偏离预设结构,则减弱甚至丧失给定的功能。因此在设计过程中结合设计后新酶的折叠状态,相比不考虑再去实验验证筛选(几千几万条),在时间和实验成本上都占有优势。然而,折叠后的构象,实际上就是蛋白质结构预测的目标。结合第二部分中对蛋白质结构预测工具的梳理,可以看到在人工智能强大的拟合能力帮助下,最近几年来在蛋白质结构预测方面获得了突破性的进展。许多蛋白质结构预测工具由于预测的高效快速被广泛应用,例如trRosetta[16]、RoseTTAFold[84]等。那么,从设计酶的“可折叠性”出发,探索将蛋白质结构预测工具与现有的酶设计改造方法相结合,将会是一条有效的酶智能设计改造思路,有助于探索更为广阔的蛋白质序列空间。
在众多优秀的蛋白质结构预测工具中,不得不提AlphaFold2[14]。AlphaFold2实现了对人类蛋白组58%的准确性预测(pLDDT高于70,可信),36%的结构位置预测高可信。其与欧洲生物信息研究所(EMBL-EBI)合作建立的平台AlphaFold DB[AlphaFold蛋白质结构数据库,AlphaFold Protein Structure Database(ebi.ac.uk)],涵盖了几乎98.5%的人类蛋白。因此,本文以AlphaFold2为代表,探索如何借助蛋白质结构预测工具增加酶设计改造的准确性。其他结构预测工具,可以根据具体研究的数据或者任务不同,替代AlphaFold2的结构预测工作。
3.1 折叠“监督器”
考虑酶的“可折叠性”,最直观的解决办法是快速预测设计的新酶的结构,检验其是否具有给定结构。因此,第一种预测是将蛋白结构预测工具作为一个监督者,约束生成的序列具有折叠成给定结构的能力[如图1(a)][41-45,85]。这个思路实施起来的最大难点是从序列预测结构的精度限制。但是现在得益于结构预测的突破性进展,使得这种设计新酶成为可能。其基本思路是在设计序列的时候,加入一个辅助的“监督者”对于生成的序列是否可以折叠且具有给定的构象进行评分,根据得分对蛋白质序列通过基于梯度的、梯度自由的或者神经网络构造的优化方法来更新序列。通过不断重复迭代这一过程,最终得到构象约束下的收敛序列。设计序列的时候一般遵从最小能量的原则。但是,我们不清楚给定的构象就一定是设计的这条序列折叠后的最低能量构象。因此结构预测作为“监督器”实际上计算了在给定结构情况下蛋白质序列的最大联合概率。

图1 结构预测工具在酶智能设计改造中的应用方向Fig.1 Specific aspects for the application of structure prediction tools in the intelligent design and transformation of enzymes
David T.Jones[41]尝试将AlphaFold2引入固定骨架设计序列的过程中,以约束生成的序列能够折叠成给定的骨架,并且正交实验中也验证了分子动力学方法模拟的结构对AlphaFold2监督后的实验结构高度支持。其具体流程是:①生成初始蛋白序列。基于研究者之前提出的基于自回归的Transformer蛋白质序列生成模型[86]生成1000条初始序列。同时对于得到的序列用AlphaFold2预测其结构,并与要设计的骨架结构用TM-align[87]做结构比对。最后选择结构比对得分最高的那部分结构的序列为初始序列,不具有高结构置信度的序列则用丙氨酸填充。这样做的好处是保证初始的序列是可收敛的,否则可能序列太随机导致最后没办法折叠。②在序列空间中执行贪婪的半随机游走,逐步突变起始序列进行迭代的端到端设计。这里面AlphaFold2的作用有两个:一个是预测序列结构,比较与要设计结构的距离直方图损失,根据损失是否减小来判断突变序列是否合理;另一个是确定该序列中哪一部分残基位点要被突变、修改。举例来说,从起始序列出发并通过AlphaFold2预测其结构以及每一个残基的pLDDT打分(衡量每个残基的局部结构合理性)。这里,计算预测结构中的距离直方图并与要设计的骨架结构的直方图计算损失。同时,利用每个残基的pLDDT打分设置为序列位点是否要被采样的概率。得分较高代表此处残基是稳定的,反之则是下一次迭代序列设计采样的点。在下次迭代采样中,对于选定的采样位点进行饱和突变,直到距离直方图损失减小,才接受序列的突变采样。这样设置的好处是对于与要设计结构的高度匹配的序列不再改变,大量减少采样时间尽快收敛以及可能引起的负协同效应。作者在人工设计的Top7上进行测试,得到的序列结构不论是通过AlphaFold2、trRosetta还是基于片段从头折叠的方法,均被证实与要设计的骨架可能是同一种折叠。该工作应用AlphaFold2在初始序列设计上保证了与目标结构的局部高结构匹配度,同时在序列设计过程中利用AlphaFold2预测的结构与目标结构的距离直方图损失约束其设计序列保持全局结构相似性以及利用残基位点可信度增强局部残基结构稳定性。同年,S.Kashif Sadiq[42]也在bioRxiv上提交AlphaDesign工作,基本思路也是利用AlphaFold2预测的结构与要设计的骨架结构的差异来限制调整序列的优化,采用的优化函数是基于进化的遗传算法来迭代生成序列。主要差别在于该方法利用预测结构的三维坐标信息差异构建目标函数优化而不仅仅是二维的配对距离直方图约束,可能在结构约束上更加有效。而且该方法扩展了可能的设计任务的范围,设计了一些长度在32~256个氨基酸、结构稳定、从头设计且具有不同折叠的单体蛋白、同源二聚体、异源二聚体、同源低聚物(三聚体到六聚体)。Baker组[43]提出的trDesign是第一个提出将结构预测工具trRosetta应用到蛋白质序列设计中的工作,考虑的也是二维距离直方图的损失来更新梯度,更新被表示为PSSM的序列。但是受限于trRosetta利用的是二维的结构约束,在正交验证中发现基于这种反向传播的方式设计序列不能很好地对三维结构进行编码,且上述三个工作均是基于给定骨架设计序列,限制了实际设计酶的应用需求。后来Baker组提出的“幻想”(hallucination)的方法[44],不从给定骨架结构出发设计序列,而是考虑在这种目标结构缺失的条件下,是否能随机产生结构和序列。其实现是通过最大化设计序列的结构与随机背景序列的差异约束,从而约束该序列折叠后的结构具有一个典型的二维结构特性[44]。实验中设想了2000条序列,聚类后发现均可以在已有的PDB结构库中寻找到相似的折叠。实验验证的时候有62条是可溶表达的(实验验证了129条),且CD的圆二色谱和目标结构的二级结构分布吻合。相比传统设计验证的方法,仅仅129条实验验证且有48%的成功率,极大地减少了人工验证的成本和时间。但是由于trRosetta精度有限以及二维结构约束的不足,在接下来的工作中将RoseTTAFold嵌入到具有给定motif的序列设计中[45]。RoseTTAFold显示利用SE-3 Transformer预测三维结构坐标以及二维距离分布,大大提高了序列设计的准确性。在免疫相关蛋白中,成功设计出携带中和性抗体表位的蛋白以及与新冠病毒S突刺蛋白受体结合的ACE2类似物蛋白。后续提出的RFjoint,不再通过神经网络不断迭代推理以及反向传播来设计序列,而是将结构预测和序列设计两大任务结合起来,直接训练全新的模型[45]。这样的好处是减少了反向推理时间,大大降低了设计的时间成本。
总的来说,结构预测工具作为结构“监督器”,在设计过程中预测设计序列的可能结构,并利用该预测结构和目标结构的差异作为损失优化模型,使模型学习到要设计的目标结构信息,从而设计具有折叠到给定结构能力的酶序列。当然根据实际设计任务的目标不同(比如结合口袋的区域等),可以将这部分信息掩盖,在恢复序列的同时利用结构预测工具预测其结构,则同时还能约束设计的酶从整体结构环境中学习到关键的局部结构。
3.2 突变“筛选器”
结构预测工具还可以作为突变筛选器,在酶智能改造设计中作为一种辅助的结构评价指标筛选有益的或者不合适的残基突变位点[如图1(b)所示]。接下来的工作介绍还是以AlphaFold2为例。AlphaFold2输出的结果分析可以提供有关新设计的局部骨架结构的准确性和可折叠性的关键信息,指示可能错误折叠的区域,并以此评估可以减轻错误折叠的突变。
Sarel Jacob Fleishman课题组[13]提出,现有功能蛋白设计方面由于错误折叠等导致的失败使得可靠的高效酶从头设计目标仍然遥不可及,因此设计了一种改善设计蛋白中不是很合理的位置方法。该方法首先利用Rosetta进行单点突变扫描,筛选有超过5种以上降低自然状态能量突变的位置标记为“次优”位置。然后应用FuncLib集中在这些低效率酶的“次优”位置上设计突变,将催化效率提高了330倍。最后利用AlphaFold2预测的pLDDT得分和计算的RMSD标记了可能错误折叠的区域,合理规避或者重新设计不合理区域,大大提高了其催化效率[13]。这种思路类似于1.1节中讨论的根据残基在当前结构环境中的“不合理”值,判断是否要在此位点突变。该工作指出,AlphaFold2分析可以提供有关新设计的骨架结构可能的准确性和可折叠性的关键信息,指示可能错误折叠的区域,并评估旨在减轻错误折叠的突变。
在设计领域,有工作通过引入pAE等来自AlphaFold2的结构指标作为“筛选器”,为4个靶点受体蛋白设计了2万条伙伴(binder)序列,并且做了相应的实验合成[88]。最后发现基于pAE指标相比传统的Rosetta打分,筛选后的序列成功率在IL2RA以及LTK靶点上数量差异分别达到了8倍、30倍。这一数量变化证明了利用结构预测工具作“筛选器”的有效性。
3.3 结构“分析器”
结构预测工具还可以作为一种辅助的结构分析,从预测的结构上分析其背后存在的催化机理,结合特异性等[图1(c)右]。通过分析突变体结构(AlphaFold2预测)与底物结合的复合物结构,来检验突变策略是否合适[91-94]。Martin Bartas则利用AlphaFold2成功预测蛋白质结构库,通过结构相似寻找具有Zα结构域(高结构保守,与Z-DNA/ZRNA结合形成)的蛋白结构[95]。这种蛋白在相关文献报道中仅有8个,但是由于AlphaFold2对蛋白结构库的丰富,发现了185个推定可能有该结构域的蛋白质结构。Xin Fengjiao课题组[96]利用AlphaFold2预测出酶序列的高精确度的合理结构,从结构角度上分析其催化性能,在与底物结合口袋附近的位点上进行合理突变,发现了高催化效率和或底物偏好性扩大的突变体。
还有研究工作利用AlphaFold2研究不同的构象变化[图1(c)左]。AlphaFold2虽然在单体结构上训练,但是可被成功应用在多肽与蛋白质的复合物结构预测中[97]。因此,合理推断AlphaFold2学习到了蛋白质在功能改变过程中构象的动态集合或者是由于突变导致的构象改变。有工作利用不同深度的MSA输入到AlphaFold2中去研究这种构象的异质性(conformational heterogeneity)[89]。Guillem Casadevall提出了一种新的观点,将基于AlphaFold2的新模板策略结合分子动力学模拟,发现不同突变的色氨酸合酶的β亚基(TrpB)结构域的一些不同闭合模式[90]。
4 讨 论
本文从头梳理了酶改造设计在利用人工智能技术方面的一系列工作,指出现有工作中存在的错误折叠甚至无法折叠导致失败,以及设计大量序列需要人工实验验证的成本问题。同时基于现有蛋白质结构预测工具的高效快速预测性,可以作为结构“分析器”、突变“筛选器”、折叠“监督器”在设计过程中帮助提高酶的“可折叠性”。正因为考虑“可折叠”能力,设计的新酶的质量相比传统大量序列中质量较高,帮助后续的实验验证降低成本的同时又提高了成功率。值得注意的是,这里面结构预测工具与酶设计工具共同采用,结构预测工具本身只是作为辅助任务。我们在讨论结构预测工具应用的时候,是以AlphaFold2[14]为代表展开介绍的。
在介绍应用的时候,我们归纳了三种应用方式。这三种应用的前提均是认为AlphaFold2这类蛋白质结构预测工具学习到了蛋白质序列到结构的复杂关系,对蛋白质结构的全局以及局部结构预测的准确度是可信的。随着越来越多结构预测工具的开发,根据不同任务(无同源序列)、不同数据类型(α螺旋结构比例较高)等,可以将AlphaFold2替换成其他的结构预测工具。例如上面提到的David Baker组提出的RFjoint[45]采用的就是该组提出的结构预测工具RoseTTAFold[84]。
关于智能方法的引用,相比传统方法,既大大减少了采样空间的计算量,又有非常优异的计算准确度,在酶的智能合成改造中的应用是非常具有研究前景的,也是有所突破的。但同时不可忽视的是,一些问题仍然存在且限制了进一步的酶功能研究。
第一个难点是如何将酶在具体参与生物过程中的反应机制等融入到智能算法的设计中。我们知道,生物反应是十分复杂的,甚至还有一些特异性或者混杂性。如何让模型学习到这种模式或者规则,仍然是需要继续探索的问题。不过好在现有的一些工作中已经开始尝试探索。例如:AlphaFold2中更新残基配对特征的时候采用的三角乘法更新,就是从我们理解的两边之和大于第三边这种距离上的约束来限制残基对在空间上的距离,从而确保更新残基捕捉合理的结构模式。又比如RFDesign中设计免疫相关蛋白设计,那么如何将免疫相关蛋白拥有的广谱性结合能力这一先验知识加入到计算蛋白设计中呢?文中考虑结合时的受体环境,设计基于三维结构坐标的能量项来表示吸引力、排斥力以及具有的球形形状三种特性。
第二个难点是对于深度学习模型来说,从海量数据中挖掘模式是合适的。但是现有的状况是酶的相关数据量小,没有统一的标准格式,是有冗余的。当然这也与特定学科有关系。很多研究工作利用迁移学习来解决数据量小的问题,比如DeepET在大的蛋白质序列-最佳生长温度(OGT)数据集上训练模型,然后迁移到预测酶的最佳催化温度和蛋白质的熔融温度[98]。或者利用自然语言处理(NLP)中广泛使用的大规模语言预训练模型学习序列的表示,然后小数据集上微调,进行一些功能预测[21,26]。
第三是关于蛋白质设计方面的。在实际应用中,研究者希望利用深度学习设计的酶序列具有可设计且可折叠性。现有酶序列设计的精度并不高,虽然利用智能算法有效降低实验室实验测定的成本,但是设计出来的序列能否被表达、能否折叠,都是需要被重点研究的。本文探讨蛋白质结构预测工具在这方面的应用,就是希望能帮助提高可折叠性酶的设计。对于没有同源序列的酶设计结构,快速有效的结构预测是有必要的。这或许可以应用现有的单序列蛋白质结构预测工具,包括TRFold、ESMFold、trRosettaX-Single、OmegaFold等。上面的工作表明这确实是一种可行性的方法,但是仅从最后结构的约束或者评价中利用结构预测的指标表明错误折叠的区域,还是很有限的。最近David Baker团队提出的RFDiffusion,通过逐步对加了噪声的结构去噪一步步恢复其结构,提出一种新的设计可能。酶的设计不再是局限于给定结构或者给定拓扑、给定功能的描述,直接设计给定功能且可靠的酶,值得期待。
第四是针对现有酶结构数据的。蛋白质序列和结构的数量差异是非常巨大的。不管最初的目的是什么,酶设计改造最终是希望设计出一个结构从而发挥相应的功能的。借助以AlphaFold2为代表的高效快速的结构预测工具,可以大幅度扩展酶的结构数据,从而分析结构上的差异,理解蛋白功能机制。同时海量结构数据直接使从结构环境中分析残基类型成为可能。
总之,人工智能技术的突破是惊人的,如何巧妙借助这股东风的力量高效且快速解决酶改造设计的相关问题,是非常具有研究前景的。
