蛋白质复合物结构预测:方法与进展
2023-07-10黄鹤吴桐王闻达李佳珊孙黛雯叶启威龚新奇
黄鹤,吴桐,王闻达,李佳珊,孙黛雯,叶启威,龚新奇,
(1 中国人民大学数学科学研究院,北京 100872; 2 北京智源人工智能研究院,北京 100084)

细胞中的大多数蛋白质与其他蛋白质或其他大分子(如DNA或RNA)结合形成蛋白质复合物,在许多细胞过程中发挥着关键作用。在分子和细胞水平上描述这些相互作用的三维结构和功能,并阐明基本的物理原理,仍然是生物学和医学的一个重要目标[1]。X射线、高分辨率核磁共振和冷冻电子显微镜解析蛋白质复合物结构既昂贵又耗时,因此通过计算手段预测蛋白质复合物的结构是非常重要和必要的。为了评估当前蛋白质结构预测算法水平,CASP(Critical Assessment of protein Structure Prediction)和CAPRI(Critical Assessment of PRedicted Interactions)比赛评测每个参赛队伍预测的蛋白质单体或复合物的结构,从开创至今已成功举办多届,促进了蛋白质结构预测的快速发展。在CASP14比赛中,由DeepMind团队开发的AlphaFold2[2]实现了高精度的蛋白质结构预测,他们设计的模型根据氨基酸序列可以准确预测蛋白质三维结构,其中大部分单体蛋白质预测的结构可以接近实验精度,这是蛋白质结构预测领域的重大突破,也为蛋白质计算领域其他问题提供了新思路。
在CAPRI第50轮比赛中[1],一共有12个多聚体题目,其中4个题目对于整个组件或主界面具有良好的结构模板,其他的只有部分亚基有较好的模板。25个小组(包括服务器)参与了CAPRI结构预测,表现最好的小组大概有70%~75%题目做到了可接受的水平,但高质量的模型较少。在2022年举办的CASP15比赛中,有87个组参加了蛋白质复合物结构预测赛道,其中一共有47个题目,大部分是蛋白质低聚物,也有超过10条链的超大复合物,对于一些蛋白质低聚物题目能够预测出较高质量的模型,但是有一部分复合物结构的预测结果不理想,准确地预测蛋白质复合物结构仍然是一个挑战。
在蛋白质复合物结构预测的相关研究中,早期的工作主要利用实验信息和生物背景知识来协助蛋白质结构预测,例如小角度X射线散射实验数据、交联数据等信息可以作为先验知识来协助构建复合物结构。如果知道某个残基对间的距离或接触信息,这有助于筛除出计算过程中产生的噪声模型(decoys)。还有一些工作开发一些文本挖掘的方法用于搜索文献中的生物信息来协助建模过程[3]。另外,打分函数用于挑选高质量的模型,它是蛋白质复合物结构预测流程中非常重要的一部分,其中涉及的物理力场和各类能量项是根据生物经验知识总结的。此外,基于蛋白质共进化思想,从多重序列比对(MSA)中获取共进化信息,通过共进化分析的思路来预测蛋白质残基间相互作用信息,这也有助于提高蛋白质结构预测算法水平。
后期的研究工作聚焦于利用人工智能算法来进行蛋白质复合物结构建模,如结合共进化分析和深度学习的蛋白质残基接触预测,促进了蛋白质结构预测领域的快速发展。后续的工作进一步地研究残基距离矩阵、二面角等几何信息预测。这些算法也被扩展到蛋白质复合物链之间的残基接触预测。其次,端到端的结构预测算法实现了高精度的单体蛋白质结构建模,这也正成为蛋白质复合物结构预测的主要手段之一。随着预训练大模型的发展,从监督学习转变为自监督学习,预训练模型的范式也在影响着蛋白质结构预测领域。
这篇综述总结了蛋白质复合物结构预测的相关计算方法。首先,我们介绍了基于人工智能算法的蛋白质结构预测方法,其中包括四个方面内容(共进化分析、残差网络与接触预测、基于Transformer的端到端结构预测方法和蛋白质预训练模型),它们之间的关系如图1所示。另外,本文也重点总结了蛋白质链间接触预测的各种思路和方法,最后介绍了蛋白质分子对接和端到端的蛋白质复合物结构预测进展。

图1 基于人工智能算法的蛋白质结构预测Fig.1 AI-based methods for predicting protein structure
1 基于人工智能算法的蛋白质结构预测
在生物信息学中,蛋白质结构预测是一个突出的研究热点,其中大量的工作聚焦于残基间几何信息预测(残基接触或距离图,朝向夹角等信息)。由于蛋白质折叠成3D结构是由其天然状态的相互作用氨基酸决定的,预测蛋白质残基之间的接触一直是主要被研究的子问题。其中基于共进化分析和深度学习的方法极大地提升了蛋白质残基接触及结构预测水平。近两年来,蛋白质单体结构预测水平已经提升到了与实验精度相当的水平,主要得益于端到端的模型,如AlphaFold2。同时还有一些基于蛋白质序列或结构的预训练模型,在蛋白质结构预测或者相关任务上也展现了不错的性能。在本节中,将围绕蛋白质几何信息预测,基于Transformer的端到端蛋白质结构预测方法和基于蛋白质序列和结构的预训练模型三个方面展开。
1.1 蛋白质几何信息预测
1.1.1 蛋白质共进化分析与残基接触预测
受自然选择影响,当具有相互作用的残基对中的一个残基发生突变时,另一个残基也会发生与之对应的突变,这种一对残基共同变异的现象被称为“共进化”。常用的共进化方法主要分为两种:第一种是假说蛋白质序列近似服从高维的正态分布,因此利用逆协方差矩阵(inverse covariance matirx)来表征残基间的共进化程度[4];第二种是假设蛋白质序列可由一个马尔科夫随机场模型(Markov random field,MRF)产生,进而采用两体项(two-body)来表征残基间的共进化程度[5-6]。
蛋白质残基间接触或距离预测是蛋白质结构预测的子问题,因为直接预测蛋白质结构三维坐标比较困难,所以先预测蛋白质的接触矩阵,然后作为约束来优化蛋白质折叠,相对来说更简单。由于距离预测较困难,前期的相关研究主要关注于残基接触预测[7-9]。当两个残基Cβ(或Cα)原子之间的欧氏距离小于0.8 nm时,则认为这两个残基具有接触(contact),否则认为没有。
早期的共进化分析通过无监督的方式考虑了残基对之间的关联关系。其中一些方法对所有残基位置建立全概率模型,再试图去除间接关联的影响,从而避免局部模型的缺陷。另一些模型通过马尔科夫随机场(MRF)对多重序列比对进行建模,从而学习一组相似序列的共进化信息,这种方法一般被称作直接耦合分析(direct coupling analysis, DCA)[5],其对于多重序列比对建模。其中马尔科夫随机场的参数可以通过极大似然法进行估计,但是由于涉及到配分函数的计算,计算相对困难,因此发展出多种近似求解方法,主要包括置信传播算法(bpDCA[10])、平均场近似算法(mfDCA[5])、系数逆矩阵协方差(PSICOV[11])、伪似然最大化算法(plmDCA[12])和混合似然算法(clmDCA[13])。其中,伪似然最大化算法是无监督的残基接触预测最先进的方法之一,最具代表性的方法是Gremlin[14-15]。Gremlin将序列简化成全连接图,用一阶项(代表残基的保守性)与二阶项(代表残基间相互作用)来计算序列的整体能量。Gremlin假设MSA中的每一条序列都存在相似的三维结构,根据玻尔兹曼分布定律,这些序列都应该具有较低的构象能量,因此训练出一套能量函数,使得蛋白质家族内所有同源序列能量最小,来获取共进化信息。Gremlin大大提升了链内残基接触预测水平,此外它对复合物链间残基也能进行相互作用预测。
1.1.2 基于深度残差网络的蛋白质几何信息预测
蛋白质结构的关键拓扑信息(如早期的残基接触预测,到后期的距离信息,键角或二面角信息等)[16-17]是蛋白质结构从头预测的最重要特征之一。经典的蛋白质结构预测是利用多重序列比对进行共进化分析,如CCMPred[18],再采用深度学习模型进行蛋白质几何信息预测,最后通过优化手段得到蛋白质结构。2016年许锦波等开创性地将深度残差网络(ResNet[19])架构成功地应用到结构预测领域中[16],显著提升了蛋白质残基接触预测,在这个工作基础上有大量结合共进化和深度学习的算法出现[7],其中代表性的工作如AlphaFold[20](主要引入残基距离预测)和山东大学杨建益等开发的trRosetta[17](主要引入了二面角信息等),均采用了深度残差网络。此外,清华大学龚海鹏团队设计的AmoebaContact[21]使用了一种新的网络架构来学习残基接触图;中科院计算所的卜东波团队开发的CopulaNet[22]尝试直接从多重序列比对中学习共进化信息,密歇根大学张阳团队开发的C-I-TASSER[23]、C-QUARK[24]结合了经典的蛋白质结构预测算法和蛋白质接触图,这一系列工作推进了蛋白质结构预测算法的进展。后续有一些工作尝试直接预测残基距离矩阵[25-26]。在通过预测残基对间的接触图、距离图、朝向夹角等信息后,通常的做法是将其转为几何势能,并利用Rosetta[27]、CNS[28]或梯度下降算法[21]使得结构势能最小,从而建立蛋白质三维模型。
1.2 基于Transformer的端到端蛋白质结构预测方法
2020年,AlphaFold2在蛋白质结构预测竞赛CASP14中大获成功,其中在AlphaFold2复杂的结构框架和运行流程中,大大小小的计算模型层出不穷,包括多重序列比对数据库构建、训练集测试集构造、特征提取计算等,此外成对信息提取、3D结构建立等模块大量使用最先进的深度学习模型。其中的核心模块是基于Transformer[29]的Evoformer,Transformer由Google团队提出,完全摒弃循环网络结构而只使用Attention机制和前馈神经网络进行神经机器翻译,Evoformer借鉴了自注意力(self-attention)机制、位置编码(positional encoding)等经典模块,并设计了三角更新(triangular update)和三角注意力(triangular attention)等模块。
此前的结构预测算法(包括AlphaFold1[20])通常是是先通过共进化分析来预测接触或距离矩阵、二面角信息,再来优化蛋白质折叠过程。2019年AlQuraishi等提出的RGN[30]模型是首个端到端的蛋白质结构预测模型,但是其模型的精度不及经典的“两步走”结构预测算法。2021年AlphaFold2[2]成功地实现了高精度的端到端蛋白质结构预测算法。总体来说,AlphaFold2的端到端结构预测算法,其模型中并没有完全抛弃几何约束等信息,而是将其作为了一个损失函数项融入到了整个模型优化过程中,最终训练好的模型也学到了较好的几何信息。端到端算法的一大优势是可以避免预测接触矩阵的误差累积到最终的三维结构,此外直接基于多重序列比对进行操作,也能够避免共进化分析带来的噪声信息。
1.3 基于蛋白质序列和结构的预训练模型
预训练模型是先在一个原始任务上预先训练出一个大模型,此模型可以用来提取一些表征信息,再针对特定的下游任务进行微调,从而可以提高在目标任务上的性能,这是现在很多领域(包括自然语言处理和视觉模型等)一种通用的模型框架。在蛋白质领域,近年来也有一些预训练模型相关工作出现。在研究蛋白质结构的领域,序列数据和结构数据是两类重要的数据表征形式。其中大部分结构数据主要是从实验室做实验获得,精度很高,但是耗时耗力,而蛋白质序列数据(很多都没有对应的实验结构数据)的获取相对容易,序列数据远远多于结构数据。早期的蛋白质预训练模型是基于蛋白质氨基酸序列数据,并应用到蛋白质结构建模或者蛋白质功能预测相关任务。此外,随着AlphaFold Database[31]和ESMFold Database[32]数据库的出现,有大量的高置信度的蛋白质预测结构,可作为RCSB PDB实验结构数据库的补充,近期也有一些工作研究直接从蛋白质三维结构出发进行预训练,在蛋白质或者小分子的相关任务上取得了不错的效果。
1.3.1 基于蛋白质序列的预训练
基于蛋白质序列的预训练模型又称为蛋白质语言模型(protein language model, PLM),是将自然语言处理领域的预训练思想应用到蛋白质建模中。氨基酸序列可以看作是一类语言,测序技术成本的降低使得我们能够获取大量天然蛋白质序列,由氨基酸序列组成的数据库在某种程度上可以视为一种语言数据库,从而可以使用针对自然语言开发的大模型对它们进行建模。
蛋白质序列预训练的模式通常是采用类似BERT[33]的模式,其核心思想是对于氨基酸序列随机遮挡一些氨基酸位置(如15%),模型旨在预测这些缺失的氨基酸。在这个模型构建中,不需要利用多重序列比对信息,也不需要结构信息作为标注,网络在学习预测氨基酸种类过程中也隐含地获得了表征信息。这些表征信息经过简单的监督学习或者回归拟合[34],可以用来做结构类的任务,如二级结构预测、残基接触预测、功能预测等。
Meta团队开发了一系列蛋白质语言模型,其中ESM[35]是第一代模型,它基于单序列(singlesequence)进行预训练,并在残基接触预测问题上取得了与经典方法相当的水平。其团队后期开发的MSA Transformer[36]考虑在 MSA上进行建模,在模型细节方面,拓展了attention机制,对MSA矩阵的行与列分别计算注意力,分别代表对不同氨基酸序列的关注程度以及对不同残基位置的关注程度。此外将mask语言模型应用于MSA中,增加预训练的难度以提高模型鲁棒性。在模型复杂度方面,MSA Transformer模型相比原本的蛋白质语言模型,模型参数量明显减少,节约了大量训练空间和算力。在训练效果方面,MSA transformer作为预训练模型,可以完成许多不同的下游任务,以无监督残基接触预测任务为例,相较于传统的结构预测模型和基于单序列的蛋白质语言模型,MSA Transformer效果显著优化,尤其在MSA深度不足时仍可以保证一定的预测准确性。
近期,结合单序列蛋白质语言模型和结构预测模块的算法,在一些孤儿蛋白或者人工设计蛋白上展示了不错的结构预测性能,如ESMFold[32]、HelixFold-single[37]、OmegaFold[38]、trRosettaXSingle[39]、RGN2[40]。当MSA质量相对较高时,基于AlphaFold2的相关模型能够保证较高的准确性,但是当同源序列比较少或者找不到同源序列的时候,基于MSA的相关算法经常得不到合理的模型。几个方法的共同之处是使用了蛋白质语言模型的表征信息替换掉原始MSA的输入信息,并采用了类似AlphaFold2的Evoformer模块和结构模块。除了在MSA质量较低的蛋白质上展示了更好的预测性能外,基于PLM的结构预测模型通过利用表征信息就能生成结构,而不需要进行MSA的构建,因此可以大大加快蛋白质结构预测的速度。
此外,还有一些工作研究基于蛋白质语言模型来预测蛋白质突变,比如ESM-1v[41]、ProtT5[42]。Ntranos团队[43]则更直接地使用单序列语言模型,分析了人类基因组中的所有蛋白质,对约4.5亿个可能的错义突变影响进行了预测,并在致病性突变预测、深度扫描突变分析和异构体特异性预测等问题上展示了可能的潜力。
1.3.2 基于结构的预训练
蛋白质结构是在三维空间中表示的,有一些工作尝试直接从三维信息出发构建预训练模型,其中能获得非常有效的蛋白质结构表征信息。与蛋白质语言模型的相似之处是不需要额外给出标签信息,蛋白质结构预训练模型仅基于结构本身的信息构建自监督学习任务,如残基对的链接信息掩码、对残基对种类进行随机删除、替换或者残基坐标加上噪声。在预训练好的模型中提取一些结构表征信息,也可以用于功能预测、蛋白质结合力预测或相互作用预测等任务。
由Guo等[44]提出的自监督的预训练模型,其思路是从蛋白质三级结构中学习结构表征信息。考虑天然的蛋白质结构受到随机噪声的干扰,预训练模型旨在估计受扰动的3D结构的梯度。该工作中采用SE(3)等变特征作为模型输入,并在保留SE(3)等变性的情况下重建3D坐标上的梯度。这种范式避免了使用复杂的SE(3)等变模型,并显著提高了预训练模型的计算效率。其预训练模型在蛋白质结构质量评估(QA)和蛋白质-蛋白质相互作用(PPI)位点预测这两个下游任务上都表现出不错的预测精度。
Tang Jian团队[45]开发的基于AlphaFold Database[31]数据库的蛋白质结构预训练模型,其中使用了约80万个数据样本。他们设计了一种简单有效的编码器(GearNet),通过添加不同类型的序列边或结构边来编码结构信息,并对蛋白质残基之间进行相关信息传递。其中采用了多视图对比学习来进行预训练,其目标是对齐来自同一蛋白质的不同视图的表示,同时最小化来自不同蛋白质的视图之间的相似性。文中使用了残基类型预测、距离预测、角度预测、二面角预测四个自监督学习任务来预训练蛋白质图编码器。其最终实验结果表明,模型在功能预测等任务上达到了与最先进的基于序列的预训练模型相媲美甚至更好的结果。
深势科技团队发布了首个三维分子预训练模型Uni-Mol[46]。Uni-Mol首先在利用2亿个分子三维构象和300万个蛋白候选口袋数据构建了预训练数据集,在进行预训练后,Uni-Mol在分子构象生成、蛋白-配体结合构象预测等三维构象生成相关的任务上取得了非常好的性能。其中蛋白质口袋预训练数据集来自蛋白质数据库[RCSB PDB(http://www.rcsb.org)],库中有约190K的结晶真实蛋白3D结构,该团队在此基础上构造一个由320万个候选蛋白口袋组成的3D构象数据集。Uni-Mol共使用三种不同的自监督策略进行模型训练:与BERT类似,Uni-Mol中也使用了对原子掩码的预测任务,采用了预测原子类型的策略;此外,使用了去噪策略,预测被掩码的原子对的欧氏距离以及直接预测被掩码的原子的正确坐标。
2 蛋白质复合物链间残基接触预测
蛋白质结构的关键拓扑信息(如残基间接触或距离信息,二面角信息等)对于指导蛋白质3D结构预测是至关重要的,其中结合共进化分析和深度学习的方法极大地提高了单体蛋白质(链内)残基接触预测,最近有一些工作尝试将链内残基接触算法拓展到蛋白质复合物(链间)接触预测。链间残基接触的定义与链内的残基接触定义类似,即残基间距离小于某个阈值(cutoff)的这对残基即判断为接触,对于距离的定义稍有不同,其中单体链内的残基间的距离是指Cβ(或者Cα)原子之间的距离,此外,复合物链间的残基间的距离也可以用最小重原子距离来表示,当一对残基的最小重原子距离小于0.6 nm(或者0.8 nm)时,这对残基即是接触的。本节从复合物序列比对拼接方法及复合物链间接触预测这两个方面展开。
2.1 复合物序列比对拼接方法
复合物的序列比对通常的构建包括两个步骤:首先是对于每条序列寻找MSA;其次是对于多个MSA进行拼接。目前最常用的多重序列比对拼接方法(MSA pairing),分别是基于基因组距离和基于进化树。EVcomplex[47]、Gremlin Complex[15]通过假设相互作用蛋白对的遗传距离小于某一阈值来配对MSA,然后基于统计模型对链间残基进行共进化分析,以预测链间残基接触。此外,许锦波等[48]提出了基于基因组信息来对MSA进行配对,这对于来自原核生物的蛋白质也有不错的建模性能。
最近有工作尝试对于拼接的多重序列比对进行打分和排序,如通过注意力机制对拼接的MSA进行打分排序[49],从而提升复合物结构预测的精度。在CASP15公布的单体及多聚体复合物结构预测算法中,郑伟等[50]在AlphaFold2使用的序列比对数据库基础上额外使用了其他数据库,增加了单条链的序列比对的多样性,此外设计了新的多重序列比对拼接策略,最后通过AlphaFold2(或者AlphaFold Multimer[51])预测的置信度分数(如plDDT)对于MSA进行打分排序。
2.2 复合物链间接触预测
早期的复合物链间接触预测主要是相互作用残基对预测[52],例如图2所示,即主要在于评测打分较高的这部分残基对(如前5、10、50)是否组成界面,高置信度的相互作用残基对预测对于研究一些结构生物机理是关键的信息,另外较准确地预测相互作用的残基对于蛋白质对接等蛋白质复合物结构预测方法也是很有帮助的。本文作者课题组开发了基于概率模型、机器学习以及深度学习等一系列的蛋白质复合物相互作用残基对预测的方法。首先,考虑到界面残基与非界面残基的区别表现在物理、化学和结构性质上,发现利用计算和统计的方法对相互作用残基进行预测是可能的[53]。在以往的研究中,提出了表面残基的许多性质,如保守性、氨基酸偏好性、疏水性、溶剂可及性等,在总结上述知识的基础上,本文作者课题组提出对残基进行了三种几何表征,即残基与其他残基的外部接触面积(ECA)、外部空面积(EVA)和内部接触接触面积(ICA)[54],并使用了统计模型对残基对进行打分,开创性地展开了对相互作用残基对预测的研究。在此基础上,提出了融合多种机器学习方法的蛋白质相互作用残基对预测方法[55]。
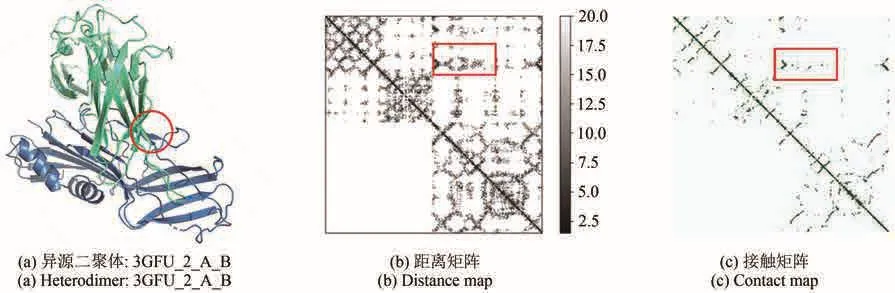
图2 蛋白质链间残基接触Fig.2 Interactions between residues with the inter-chains of proteins
此外,本文作者课题组基于长短期记忆网络(LSTM)的方法来预测异源二聚体、三聚体和四聚体的相互作用残基对[56-59],在该系列方法中,充分利用几何特征来描述残基的性质,改进了LSTM方法(结合了注意力机制[58]、图神经网络[57]、结合卷积神经网络和支持向量机的方法[60]等),并在三聚体和四聚体等多聚体数据集上进行测试,为研究多聚体复合物结构预测相关算法提供了新的思路。
基于残差网络的残基接触预测算法RaptorX-contact[16]在2016年CASP12中获得残基接触预测赛道第一名,展示了深度学习方法在蛋白质残基接触预测方面的高效性能。在2018年许锦波团队将RaptorX-contact应用到异源二聚体的链间残基接触预测(RaptorX-Complex Contact[48])。该方法沿用了单体残基接触预测的训练集和模型,仅对输入的两条链的多重序列进行拼接,即输出了异源二聚体的接触矩阵,这也是首次成功用于链间接触预测的深度学习方法。此后,在2021年该团队结合了蛋白质语言模型的信息以及原子、残基和表面的特征,最后通过残差网络预测异源二聚体的接触矩阵(Glinter[61])。程建林团队设计了几种算法来研究异源二聚体的残基接触预测,其中一种思路是基于几何深度学习算法DeepInteract[62],其中使用了蛋白质的几何信息(以蛋白质单体的结构信息为基础),另一种思路是基于注意力机制的方法CDpred[63],其中基于单体距离矩阵、共进化分析和蛋白质语言模型等特征,并使用了自注意力机制。这两个工作将异源二聚体的接触预测提升到了较高的水平。
华中科技大学黄胜友课题组提出了系列算法DeepHomo[64]、DeepHomo2.0[65],来研究同源二聚体或多聚体的链间残基接触预测问题,其中使用了单体结构信息、共进化信息以及来自分子对接的特征。DeepHomo2.0额外使用了蛋白语言模型的表征信息,此外程建林等提出的DRcon[66]也使用了蛋白质语言模型的表征信息。本文作者课题组开发的PGT[67]使用了图注意力网络并借鉴了AlphaFold2中的三角更新模块,在同源二聚体链间接触预测问题上有不错的性能。与异源复合物的残基预测水平相比,同源复合物的残基接触预测精度更高,这也与复合物结构预测中的结论相一致,即同源复合物的预测水平比异源复合物更高。
由于在复合物序列比对拼接过程会引入一些数据噪声,不使用拼接的多重序列比对也是一种选择,如只基于单体的MSA的蛋白质复合物结构预测算法[68]有不错的性能。本文作者课题组提出的基于图像修复的方法PDII[69],只使用链内的接触矩阵,而不需要使用多重序列比对信息,仅从蛋白质链内的相互作用信息来学习链间接触。基于图像修复的模型不需要使用拼接的多重序列比对数据,仅把单体蛋白质的内部接触图拼接作为模型的输入,也不需要提取其他物理化学特征;其次,这个模型不依赖于输入结构的形式,模型对于bound或者unbound的蛋白质结构输入也具有强鲁棒性;另外,此模型可以处理同源二聚体或者异源二聚体。上述的复合物链间接触预测方法均总结于表1中,包括使用的输入特征、网络架构和任务等。
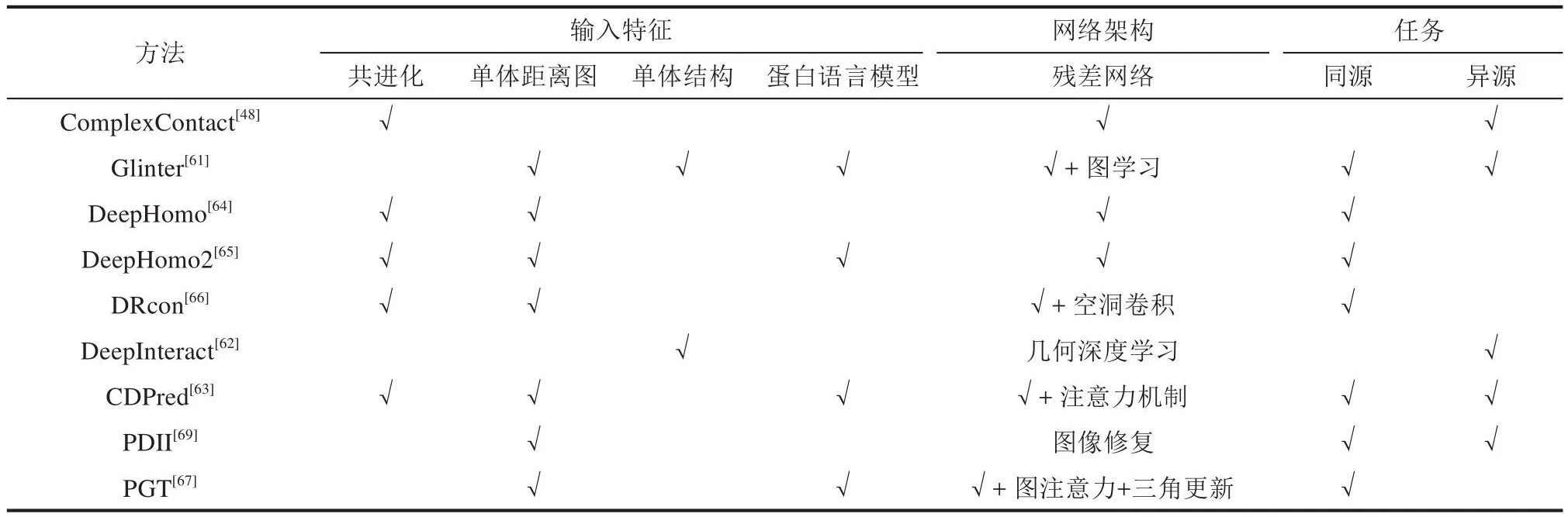
表1 蛋白质链间相互作用预测方法[48,61-67,69]Table 1 Overview of methods for predicting interactions between the inter-chains of proteins[48,61-67,69]
在考虑多聚体复合物的残基接触预测问题中,首先需要判断的是两条链是否有相互作用[52]。本课题组使用了一种基于网络蛋白质相互作用(PPI)的预测方法Sim[70],该算法从蛋白质相互作用界面的互补性和基因复制两个角度设计,可以挑选出更容易相互作用的蛋白质对。此外,课题组考虑了基于清华大学丘成栋课题组[71]开发的自然向量法,首次来预测PPI中非相互作用的蛋白质对[72]。将预测两个蛋白质相互作用或者非相互作用结合起来,这可以减少通过实验来确定蛋白质相互作用的时间和经费消耗。此外,浙江工业大学张贵军课题组[73]近年来的一些工作研究多域(multi-domain)蛋白质的相互作用,其中Sen等[74]通过研究蛋白质数据库发现蛋白域之间相互作用和链之间的相互作用有相似之处,能否借助多域蛋白质的数据来提升蛋白质复合物的结构预测也是一个值得关注的方向。
目前的端到端的结构预测算法性能(AlphaFold2)超过了传统的“两步走”的结构预测算法,因此旨在提升蛋白质残基接触预测精度对于单体蛋白质而言意义不大。但是通过研究残基接触信息预测来进一步探索蛋白质复合物结构预测相关问题仍然是有意义的,一方面是链间残基接触预测信息可以为结构生物学研究问题如蛋白质功能的研究提供先验信息,另一方面是端到端的结构预测方法如AlphaFold Multimer的预测精度相对较低,因此提升链间残基接触矩阵预测性能是有研究价值的,也很有挑战性。
3 蛋白质复合物结构预测算法
经典的蛋白质复合物结构预测算法通常是基于蛋白质分子对接方法,随着单体蛋白质结构预测算法的发展,端到端的蛋白质复合物结构预测算法也展现了不错的性能,其中大体思路是类似的,首先是根据多重序列比对和模板搜索来构建特征,其次是设计一个监督学习框架来搭建从序列到结构的端到端的算法。相比较蛋白质单体结构预测精度,蛋白质复合物结构预测算法还是相对较低的水平。在这里主要讨论的是蛋白质复合物结构预测算法建模的相关算法,本节围绕着链间接触矩阵与复合物结构预测、蛋白质分子对接以及端到端的复合物结构预测三个方面展开。
3.1 链间接触矩阵与复合物结构预测
前文系统地总结了链间接触预测的主要方法,这里简单地介绍基于预测的链间接触或者残基相互作用预测,来构建复合物结构。如Rosetta[27]、CDPSP[75]、GDFold[21]是针对单体结构预测开发的算法,其中DeepComplex[76]和DRLComplex[77]是针对蛋白质复合物开发的基于接触矩阵的蛋白质结构预测方法,主要研究如何从精度较高的接触矩阵来构建准确的3D结构,此外由Baek等[78]在CASP14提出的算法也使用了链间接触预测作为指导来对复合物结构进行建模。其中Rosetta是经典的依赖于能量项的建模,可以将置信度较高的残基接触转换成约束,来指导蛋白质折叠过程。CDPSP通过蛋白域之间的接触预测来优化多域蛋白质的结构。DeepComplex同样采用的是类似GDFold的梯度下降算法,来对复合物进行结构建模。而DRLComplex是采用强化学习的思想来优化蛋白质复合物结构。
高置信度的相互作用残基对可以为构建复合物结构提供重要的约束信息,像传统的单体结构建模过程中通过预测链内接触矩阵来限制优化蛋白质的折叠过程一样,研究者也在多聚体建模时通过预测链间的接触矩阵或预测哪些残基会相互作用来限制蛋白质复合物的建模过程[64]。此外,预测的相互作用残基对可以作为先验信息,来指导蛋白质分子对接过程。
3.2 蛋白质分子对接
蛋白质分子对接方法的流程是基于已给定的结构来预测复合物结构。蛋白质对接的思想来源于锁钥模型和诱导拟合理论。对接过程应获得同时满足空间形状互补和能量最小化原则的最佳结合模式。传统的对接过程一般通过快速傅里叶变换(FFT)、蒙特卡洛、遗传算法等方法搜索构象空间,获得大量候选蛋白质复合物构象,然后通过评分函数对这些构象进行排序和选择,最后根据能量模型对预测的结构进行优化。下文描述了一些对接算法,包括能够输入多个亚基的方法、专门针对具有对称性的同源寡聚体的方法、基于深度学习的分子对接方法。
一些服务器能够通过输入两个以上的亚基来为蛋白质复合物建模。例如HADDOCK[79],一种用于建模多聚体的灵活对接算法,它以模糊的相互作用约束(AIR)编码来自已识别或预测的蛋白质界面的信息,以驱动对接过程。与HADDOCK不同,Multi-LZerD[80]不需要限制额外的生物信息来建模多聚体,首先生成成对对接预测,然后使用遗传算法探索构象空间,最后基于蒙特卡洛优化预测结构。这两个服务器能够对两个以上链的多聚体进行建模,并且不限于具有对称性的同源多聚体。
有一些服务器专门为具有对称性的同源多聚体结构建模,复合物的蛋白质结构主要有两种对称性,环状(Cn对称)和二面体(Dn对称)。例如,SAM[81]、HSYDOCK[82]分别为Cn对称和Dn对称的多聚体建模,Galaxy[83]系列中也针对Cn和Dn采用不同的策略GalaxyTongDock_C和GalaxyTongDock_D。此外,SymDock2[84]、MZDOCK[85]等也支持对具有Cn对称性的低聚物进行建模。
还有许多二聚体对接方法,大多数对接程序基于快速傅里叶变换(FFT)对整个构象空间进行全局采样,例如ZDOCK[86]、pyDock[87]、ClusPro[88]、MDOCKPP[89]、CoDockPP[90]、GalaxyTongDock[83]等,并且如果受体或配体在相互作用时发生大的构象变化,则建模质量大大降低。也有一些基于能量优化的随机搜索算法用于对接过程,例如,RosettaDOCK[84,91]基于蒙特卡洛搜索方法,该方法擅长蛋白质局部构象探索,但不擅长全局对接。整个过程的计算效率很低。在使用RosettaDock进行对接之前,通常使用其他刚性对接软件进行初步构象探索,并选择几个合理的构象作为起点。SwarmDock[92],基于粒子群优化算法来寻找蛋白质相互作用的低能量位置和方向。此外,还有一些其他的对接方法,例如LZerd[80],它使用3DZD来表示蛋白质界面,是旋转不变的,并基于几何哈希方法找到候选姿势。Baker等[78]在CASP14中采用了一种新的同时折叠和对接的方法,基于梯度能量最小化来采样结构。链间接触预测的质量对于这种方法很重要,随着基于机器学习的链间接触预判和距离预测方法的进步,这种方法可以大大提高对接准确率。
在CASP-CAPRI竞赛中,许多小组在建模多聚体时采用了结合模板建模和自由对接的方法[1],还开发了一些集成基于模板和自由对接的服务器。例如,HDOCK集成了同源性搜索、自由对接、基于模板的建模和生物信息集成等过程,不仅支持受体和配体的结构输入,还支持序列输入。类似的混合策略有InterEvDock2[93]、CoDock[90]、GalaxyHommer[94]、GalaxyTongDock[83]等。
除了上述传统的对接算法,最近还出现了一些基于深度学习的端到端建模算法,用于蛋白质-蛋白质刚体对接。Octavian Eugen Ganea等[95]提出了一种基于成对独立SE(3)-等变图匹配网络的刚性对接算法EquiDock,它通过优化传输和可微的Kabsch算法,使用关键点匹配和对齐来逼近结合口袋,并预测对接相对位置。通过预测旋转和平移,使得配体能够相对于受体放置在正确的对接位置,无论两个结构的初始位置如何,该方法保证预测的复合物总是相同的。与传统的对接算法不同,它不依赖广泛的采样、排序、结构优化和模板,比传统对接方法快80~500倍。
由于需要在数据集上对比不同的蛋白质复合物建模方法的性能,因此需要非冗余和高质量的数据集,如Benchmark5(BM5)[96]、PPI4DOCK[97]基准集、Huang等创建的用于对称蛋白质对接的SDBenchmark[98]。其中,Benchmark5(BM5)是最常用的对接数据集。
3.3 端到端的复合物结构预测
端到端的蛋白质结构预测算法AlphaFold2极大地提高了蛋白质单体结构预测水平,因此很自然的想法是去探究这种端到端的结构预测算法在蛋白质复合物结构问题上性能如何。早期的尝试是采用了与RoseTTaFold中开发的类似技巧,将复合物的多条序列拼接后构建复合物的结构,并直接基于AlphaFold2的模型来构建复合物的结构,其中在一些蛋白质上能够预测出质量很高的模型。Mirdita等基于AlphaFold2开发的ColabFold[99],一个对用户友好的蛋白质结构预测工具,其中也使用了一些策略来对蛋白质复合物进行结构建模。之后Elofsson实验室基于AlphaFold2提出FoldDock[100]方法,使用了配对MSA的策略,基于AlphaFold2及AlphaFold multimer方法,开发并测试了一组大型基准异二聚体,之后又针对多聚体复合物进行了预测评估[101]。这些结果强调了基于AF2的方法相对于其他对接方法的优势[102]。与此类似的工作是Gao等提出的AF2Complex[68]通过填充间隙和模板作为输入,使用每个链单独的MSA,而不使用拼接的MSA,并在多个循环步骤后通过AlphaFold2生成更多的模型,最后通过重新定义的置信度分数来挑选模型 。
尽管上述方法对二聚体(或三聚体)很有效,但它们可能存在局限性,因为对于一大部分复合物,很难获得高质量的拼接MSA作为其输入,这和上节中介绍的链间残基接触预测方法所面临的问题是类似的,即模型的精度非常依赖于输入的MSA质量。
由深势公司团队开发的可训练版本UniFold-Multimer[103],性能和AlphaFold Multimer相当,其针对对称复合物再训练了一个版本UniFold-Symmetry[104],对于对称的超大复合物取得不错的建模效果。此外,同样由Elofsson实验室提出的MolPC[105],尝试对更大的复合物(超过10条链)进行结构建模,其中使用蒙特卡洛树搜索将预测的子组件组合在一起。之后Dima Kozakov等[106]将Alphafold2与Cluspro结合起来,通过ClusPro对接的前10个结果作为模板送入AlphaFold2进行微调[78,81],也得到了一些不错的模型。
基于AlphaFold2的各类复合物结构预测工作层出不穷,Deepmind团队也在AlphaFold2基础上开源了AlphaFold Multimer[51],用于端到端的复合物结构预测。其在AlphaFold2基础上主要做了如下几个修改:修正的损失函数(其中考虑了预测结构和真实结构的对应关系);构建拼接的多重序列比对;在位置编码上增加了复合物不同链的信息[89-91]。蛋白质复合物结构预测算法也能相互作用识别。后来,Baker团队[107]利用共进化分析,并结合AlphaFold2和RosettaFold为真核生物核心蛋白质复合物的结构建模[108],开发了一个识别可能相互作用的蛋白质对并为这些蛋白质复合物的结构建模的方法。该方法首先识别同源蛋白质,生成同源基因群;然后为每对酵母蛋白质对建立同源序列的多序列比对;接下来通过一个轻量型的两轨RosettaFold模型预测蛋白质对之间的接触概率,或根据实验数据识别PPI候选;最后过滤候选的PPI,用AlphaFold2为复合物结构建模。通过筛选出830万对酵母蛋白,从中识别出1505种可能的相互作用复合物,699个复合物的结构在之前的实验中被解析,同时也为其他806个尚未结构表征的蛋白质构建了结构模型,其中,700个有实验数据支持,106个此前从未被描述。
整体来说,端到端的复合物结构预测算法能预测出比较合理的复合物结构,其中在同源多聚体上的模型精度较大,但是在异源多聚体上不太理想,其原因是同源多聚体通常不需要拼接MSA,而异源多聚体获得拼接MSA难度较大。此外,端到端的蛋白质复合物结构预测算法在一些低聚体复合物或者同源复合物上展现了比蛋白质分子对接算法更好的性能,但是分子对接在与小分子相互作用的复合物建模中更有优势,对于超大的蛋白质复合物,分子对接方法可以做出较合理的模型,这是目前端到端的复合物结构预测方法很难做到的。另外,端到端的复合物结构预测算法在多肽复合物或者抗体-抗原复合物蛋白质上表现的结果欠佳[108-110],仍有大量可提升的空间。
4 挑战与展望
本文介绍并讨论了多种计算方法,首先围绕基于人工智能的单体结构预测算法展开,介绍了常用的深度学习框架和预训练模型的新范式。此外,针对蛋白质复合物结构预测中的三个方面展开介绍,如详细介绍了针对链间接触预测的算法,再从基于对接的方法到基于人工智能算法的端到端的蛋白质复合物结构预测方法。总体来说,在链间残基接触预测、蛋白质复合物分子对接、端到端的复合物结构预测三个方面,仍有未解决的问题。
链间残基接触和距离图的预测对于指导蛋白质结构预测和蛋白质对接中的复合物结构建模很重要。目前的方法针对同源二聚体或多聚体的预测性能较高,异源复合物的链间残基接触预测精度较低。拼接的MSA质量较低,也是一个挑战,目前较常用的基于进化树和基于基因组的方法有值得改进的空间。
目前,大多数对接算法考虑刚性对接,少数算法考虑柔性,但性能有待提高。刚体对接需要两个单体未结合时的结构接近结合时的结构,对于一些困难的题目,在结合过程中,当其中有一个蛋白质的结构发生了显著变化,刚性对接方法无法为它们产生高质量的对接结果。柔性对接允许一定的构象变化,可以为某些复合物提供更精确的模型,但对于比较复杂的多聚体复合物仍不能产生好的结果,因此考虑构象变化仍然是多聚体复合体预测的重要挑战。此外,大多数蛋白质分子对接算法都只考虑二聚体,对于大于两条链的复合物,一些研究人员开发了专门针对具有Cn或Dn对称性的寡聚蛋白的建模算法,但对于非同源的多聚体结构预测仍然是未来的一个重要挑战。此外,许多蛋白质对接算法考虑了整合各种生物信息,这是有助于蛋白质结构预测的,如何使用多种生物信息,也是未来的一个重要方向。
AlphaFold2和RoseTTAFold在单体结构预测上展现了非常好的预测水平,AlphaFold Multimer在复合物结构预测中也能够得到一些不错的预测结构。总体来说,不同于以往基于模板建模和从头对接的方法,AlphaFold Multimer这种端到端建模方法可能是未来的一个重要趋势。目前蛋白质复合物结构预测整体上离单体结构预测精度还是有不少差距,抗体抗原复合物、多肽复合物、无序蛋白相互作用蛋白质复合物、超大蛋白质复合物[111-112]的结构建模也是重要的挑战。有一些后续工作更加关注如何提升多聚体复合物结构预测的性能,大部分聚焦在复合物多重序列比对的采样,在最近的CASP15比赛中,有47个多聚体复合物结构,其中表现较好的参赛组在多重序列比对采样上使用了多种新策略,并依赖于或者直接使用AlphaFold Multimer来预测最终结构。
目前在蛋白质复合物结构问题预测中,仍有几个方面值得讨论:首先是在不清楚蛋白质复合物中各单体计量比的情况下,是否有可能预测出复合物组成,比如预测出某些链的相互作用可能有助于解决这个问题;此外对于异源多聚体,如何解决不同单体之间的排列顺序问题,目前AlphaFold Multimer的方案是一种解决办法;另外,对于多聚体复合物来说,多聚体复合物的模板构建仍然是个问题,传统的蛋白质模板库主要是针对单体蛋白质的,因此,蛋白质复合物模板数据库的建立也是值得关注的。
目前RCSB PDB数据库中约有20万实验解析的结构,其中的蛋白质复合物中约有11.5万(二聚体约6.3万、三聚体1.3万、四聚体2万);AlphaFold Database以及ESMFold Database中分别有2亿和6亿个蛋白质结构,但是其中仅包含单体数据。基于蛋白质结构(实验结构或者预测结构)的方法是一种有效手段来帮助蛋白质复合物结构预测。此外,借鉴预训练模型的方法来解决蛋白质抗原抗体复合物、蛋白质和小分子复合物、蛋白质与RNA/DNA的复合物等结构预测问题是一个值得研究的方向。随着多模态的算法发展,蛋白质序列数据、结构数据、分子动力学、蛋白质组学研究结果、小角散射数据以及一些其他实验相关的数据都可以作为有效信息加入到模型中。
致谢:感谢中国人民大学公共计算平台和北京智源人工智能研究院对本课题的支持。
