人工智能时代下的酶工程
2023-07-10康里奇谈攀洪亮
康里奇,谈攀,洪亮
(1 上海交通大学物理与天文学院,上海 200240; 2 上海交通大学自然科学研究院,上海国家应用数学中心(交大分中心),上海 200240; 3 上海人工智能实验室,上海 200240)

酶是一种具有催化作用的生物大分子。经过自然选择,生物体内各种各样的酶在具备一定活性和稳定性的同时还保留了底物选择性等特异性功能。然而酶的序列空间在20N(N为蛋白质一级序列长度)量级,即便是经过了千百万年的演化,自然界也只是探索了序列空间很小的一部分,这些功能还有提升的空间。因此酶工程的一项重要任务就是通过引入突变或新的结构功能域改造酶来提高特定性质以满足工业领域或实验室的需求[1]。
蛋白质一级序列中离散的氨基酸具有高度的进化相关性,因此是酶工程主要的编辑改造对象。早在20世纪中期,Lerner等[2]就已经使用化学诱变的方法在细菌中引入突变。由于这种突变的靶向性无法控制,这项工作只是定向进化领域一次具有代表性的尝试。在重组蛋白技术发展成熟之后,蛋白质一级序列中的氨基酸可以被精准控制和编辑[3]。在此基础上,蛋白质层面有关工作机理和理化性质的先验知识可以被转化成蛋白质序列设计方案。理性设计方法便是依赖这些知识判断具体氨基酸替换后是否会增强蛋白质的特定性质,或者改造蛋白质的特异性功能,但这种方法不适用于工作机理或结构未知的蛋白质[4]。定向进化策略跨越了理性设计的知识壁垒,该方法通过随机突变和高通量筛选加速蛋白质向特定指标的进化过程,研究人员不再需要了解蛋白质的结构和工作机理。之后一系列半理性设计策略结合了理性设计和定向进化两种思路,通过构建更小也更合理的突变体文库提升效率。由于酶工程的实验结果可以按照统一的标准被收集,随着实验结果的累积,大量的数据推动了数据驱动的酶工程的发展。人工智能为酶工程提供了新的工具,机器学习方法与深度神经网络在该领域得到了有效利用与发展。总体来看,酶工程经历了从知识驱动到数据驱动的发展历程,并且二者紧密结合,相辅相成。
1 定向进化与半理性设计
早期的酶工程需要通过理性设计决定突变位点,成功的案例必须建立在丰富的先验知识上[4]。定向进化技术的核心思路可以被分为两步,先构建大规模随机突变文库,再通过高通量实验筛选得到有益突变体。这样的过程往往会被迭代实施很多轮,直到有益突变位点积累到使蛋白质性质满足预期的数量。定向进化的发展让酶工程不再需要理解蛋白质的工作机理、结构或特定氨基酸替换的具体影响,这是酶工程历史上的重大突破。Frances H.Arnold因为在该领域做出突出贡献而获得了2018年诺贝尔化学奖。她和她的团队利用易错PCR技术成功实现了枯草杆菌蛋白酶E(Subtilisin E)的进化[5]。经过3轮的诱变和筛选,最终在60%的二甲基甲酰胺(dimethylformamide)溶液中得到了相比野生型提高了256倍活性的6点位突变体。另一个具有代表性意义的工作是Stemmer在1994年提出的利用DNA重组构建随机突变文库[6-7],这项技术利用PCR扩增目标蛋白的同源基因文库并将它们剪切成大量基因片段,通过无引物PCR技术重组后,基因片段会组成杂交基因并被克隆到表达载体中供后续筛选,得到的突变体会被用于构建新的DNA片段文库,有益的突变会在如此反复的筛选过程中累积(图1)。Stemmer团队使用该方法对β-内酰胺酶(β-lactamase)进行了三轮重组(shuffling)和两轮回交(backcrossing),最终得到的突变体使宿主大肠杆菌(Escherichia coli)对抗生素头孢噻肟的抑制浓度比野生型提高了32 000倍。Liebeton团队[8-9]将多种定向进化策略结合在一起来改造铜绿假单胞菌(Pseudomonas aeruginosa)中的细菌脂肪酶(bacterial lipase)。该团队先利用易错PCR技术,在多轮迭代过程中找到数个对蛋白质产物选择性影响较大的阳性单点突变体。然后在这些阳性突变所在位置进行饱和突变(saturation mutagenesis),得到了之前随机突变过程中漏选的更好的阳性突变。在这些结果的基础上,再利用定点突变技术(site-specific mutagenesis)重新设计多点突变,最终得到的突变体在特定产物的选择性上比野生型提高了23.5倍。这种将多个定向进化策略结合起来的方法降低了随机突变漏选优秀突变体的概率,同时为饱和突变技术提供了关键的氨基酸位点。
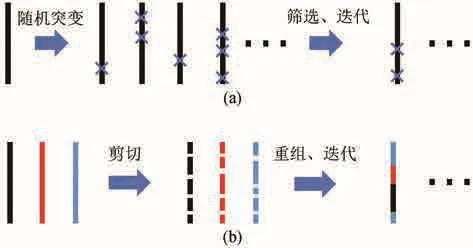
图1 易错PCR技术(a)与基因重组技术(b)的原理Fig.1 Principles for error-prone PCR (a) and DNA shuffling (b)
定向进化利用构建大量随机突变文库和高通量筛选的方法突破了酶工程关于催化机理、结构和特定突变影响的知识壁垒。然而对于许多蛋白质来说,高通量实验的设计仍然是一个挑战,并且多轮迭代筛选的方案会导致过长的实验周期,这对于生化实验室来说是巨大的负担,因此一部分研究重点被转移到了结合理性设计的小型突变体文库的筛选中。共识序列(consensus sequence)是半理性设计中具有代表性的方法[10-11]。蛋白质一级序列中氨基酸之间具有高度的进化相关性,从进化角度来看,对酶活性和稳定性产生重要影响的氨基酸很可能是保守的。在给定蛋白质家族的多重序列比对(multiple sequence alignment)中,某个位置上的保守氨基酸具有更高的频率,这些残基被认为是共识残基(consensus residue)。共识序列的核心思想是氨基酸频率反映了某些生物特性的相对重要性,在给定位置上用共识残基代替非共识残基往往能优化蛋白质性质[11]。图2以绿色荧光蛋白(green fluorescent protein,GFP)为例,展示了识别共识残基的基本原理。半理性设计是理性设计和计算方法的结合,筛选突变位点时理性思考同样重要。比如在通过酶工程提高酶的耐碱性时,序列中的天冬酰胺(asparagine)和甘氨酸(glycine)被认为对酶在碱性环境中的稳定性有负面影响,会被优先用其他氨基酸替代[12-13]。除此以外,分子动力学模拟、分子对接、第一性原理计算以及利用能量函数模拟退火等方法可从结构和能量角度筛选突变体[14]。这些计算设计方法着眼于单个或者多个突变给蛋白质结构和功能带来的具体影响,可以构建相比于定向进化更小也更合理的突变文库,大幅度降低了定向进化方案中筛选突变体的工作量,具体进行实验时不再需要高通量筛选方法。Khersonsky等[15]基于多重序列比对和能量函数提出了针对酶的活性口袋进行设计的通用方案。该方法需要研究者基于先验知识将参与突变的氨基酸限制在活性口袋内部,因为这些点位对酶的功能有更直接的影响。该团队先利用多重序列比和Rosetta能量打分过滤掉不合理的单点突变,然后对剩下的单点突变进行组合。这个方法和定向进化最大的区别在于略过了从单点突变到多点突变的叠加过程,这意味着最终表现优秀但叠加过程中表现下降的多点突变体不再会被剔除[16]。在单轮实验中测试了磷酸三酯酶(phosphotriesterase)的数十个突变体针对不同底物的水解活性,结果表明多个突变体在新的底物上表现出数千倍于野生型的活性。中国科学院微生物研究所的吴边团队[17-18]同样利用Rosetta改造天冬氨酸酶。在深入了解酶的催化机理的前提下,保持进行催化反应口袋中氨基酸不变的同时对靠近底物特异性基团的氨基酸进行突变,经过对数十个突变体进行实验测试,最终使酶在保持催化功能不变的情况下适用于多种不同底物。在定向进化中加入理性设计更有利于设计针对新底物、新功能的突变文库,且这类文库体量更小,阳性率也更高。

图2 GFP蛋白的部分序列比对结果,共识残基已被高亮处理Fig.2 Sequence alignment of GFP with most conserved residues highlighted
2 人工智能助力酶工程
蛋白质一级序列由20种天然氨基酸构成,氨基酸的离散性使蛋白质在酶工程中具备高度的可编辑性,同时在计算机中具有可编码性。除此以外,大量突变体的实验结果都能够以一种标准化的方式整合起来构成突变体数据库。这些数据推动了人工智能技术在酶工程领域的应用。
2.1 传统机器学习助力酶工程
机器学习的方法是将大量蛋白质信息按照一定方式编码,使计算机产生可以执行复杂决策的算法。Capriotti等[19]在2004年利用1615个单点突变数据训练单层感知机并预测蛋白质突变对热稳定性造成的影响,他们将测量蛋白质突变稳定性变化时的温度、pH值、单点突变内容、溶液可及性以及单点突变周围氨基酸频率分布编码并输入到模型中,使模型在预测精度上超过了之前利用能量函数计算热稳定性变化的方法。这种编码方案只利用突变周围的氨基酸频率分布将蛋白质结构信息纳入考虑,该团队在2005年推出了基于支持向量机(SVM)的I-Mutant2.0,在结构信息之外又成功编码了蛋白质序列信息[20]。曲玉辰等[21]利用I-Mutant2.0辅助设计与优化病毒融合抑制多肽,证明这种方法具备一定的可行性。早期机器学习方法使用的网络比较简单,研究重点在编码信息的选择上。编码信息应该尽量覆盖关键特征,但同时不能过度冗余导致模型过拟合等问题[22]。Fariselli等[23]同样使用基于支持向量机回归(SVMeregression)的方法来预测蛋白质突变体热稳定性的变化,但编码了更加复杂的信息从而获得比以往模型更高的精度。Laimer等[24]不仅增加了编码内容,还通过整合的方法丰富了模型架构,使用的数据包括统计模型的打分,蛋白质残基数目、二级结构、溶液可及面积、质量、亲疏水性和等电点等理化性质。这些数据会被编码输入到3个模块中,3个模块包括具备单个隐藏层的人工神经网络(artificial neural networks)、支持向量机(SVM)和多重线性回归(multiple linear regression)。经过测试,该整合模型被证明具有超越以往模型的精度[24]。
也有一些方法没有选择编码复杂的蛋白质信息,而是利用机器学习方法对现有技术进行整合互补。Dehouck等[25]选择利用多种统计势能的线性组合来预测突变带来的热稳定性变化,该方法在预测速度上相比其他方法有巨大的提升。Pires等[26]则是利用支持向量机(SVM)整合了突变体阈值扫描矩阵(mutation cutoff scanning matrix,mCSM)和定点诱变(site directed mutator,SDM)两种属性互补的方法,其中mCSM是一种利用结构特征预测错义突变(missense mutation)的机器学习方法,SDM则是一种包含了同源蛋白进化信息的统计函数[27-28]。
2.2 深度学习助力酶工程
目前人类已经从自然界中揭示了上亿条蛋白质的一级序列,这个庞大的数据库中埋藏了人脑难以理解的蛋白质氨基酸排列和进化的规律[29]。计算机硬件的飞速发展允许我们使用深度学习网络提取其中的特征信息,从而替代传统机器学习中手工提取特征的方法。蛋白质包含冗杂的理化信息,神经网络学习的对象可以是单一蛋白质的序列或者结构,也可以是整个蛋白质家族的信息。
语言模型(language model)是自然语言处理领域的核心问题,它是能够基于已有文本预测空缺处的字符或词汇的一类神经网络,能够学习某种语言的语义和语序并生成符合规则的新语句。蛋白质语言模型是这类语言模型在生物化学领域的迁移应用,它将20种天然氨基酸当作词汇,学习蛋白质一级序列中的语义和语序规则,以完成预测蛋白质功能、结构等下游任务。Facebook AI团队[30]在Transformer架构基础上开发了可以直接对蛋白质突变体进行非监督学习(unsupervised learning)的蛋白质语言模型ESM-1v。该模型使用的训练集包括9800万条蛋白质序列,使用的训练方法为随机遮掩(masked training),即输入经过随机遮掩处理的残缺蛋白质序列,令模型通过未遮掩部分来预测被遮掩部分的残基类型。这样的训练方法可以让模型具备评估蛋白质中氨基酸保守性的能力,即某个序列中特定残基类型是否符合自然界中蛋白质语言的语义和语序规则。若突变体相比于野生型更加符合模型学习到的规则,模型就会对该突变体给出阳性打分。
特定位置残基的突变受到整个自然界蛋白质语言规则的约束,也在进化过程中和邻近残基互相产生影响。MSA-Transformer模型通过编码同源蛋白质的多重序列比对(MSA)结果来学习蛋白质序列在进化过程中的约束信息[31]。该模型使用的数据库包含2600万组MSA,平均每个MSA包含1192条蛋白质序列。架构上该模型依然以Transformer为基础,但是新增了行注意力(row attention)和列注意力(column attention)两种轴向注意力机制来充分提取MSA中的信息。
蛋白质三级结构比一级序列包含更多的信息,尤其是蛋白内部氨基酸三维空间互作信息,目前通过实验解出的结构约20万条,远少于目前已知的序列数量。AlphaFold2作为深度学习模型,能够以极高的准确度根据序列预测蛋白质三级结构[32]。ESM-IF1模型使用经过AlphaFold2预测的1200万条蛋白质序列的结构进行训练,根据蛋白质骨架坐标预测其序列[33]。模型架构方面ESM-IF1使用几何向量感知机(GVP)来编码蛋白质三维结构,该模块可以保证编码信息向量的等变性以及标量的不变性[34]。Zhou Bingxin等[35]提出了轻量级的深度几何训练模型LGN,从蛋白质三级结构中学习适用于多任务的蛋白质表示。LGN在训练过程中考虑了生物学的先验知识,具体改进包括在训练过程中给氨基酸类型加入噪声来模仿自然界中的随机突变,在氨基酸节点预测的损失函数打分机制中引入标签平滑来鼓励同类氨基酸之间的置换等。LGN作为轻量级模型,其参数量和运行时间远小于同类模型,但是该模型在预测多种蛋白质多点突变fitness任务上的精度超过了ESM-IF等同类模型。
ESM-1v、ESM-IF1和MSA-Transformer等无监督模型不需要经过额外训练即可直接在特定蛋白质上执行突变体的预测任务,但打分的规则并不是蛋白质活性或者稳定性等具体指标,而是突变体相比于野生型是否更加符合模型学习到的规则。这套规则更多是在从进化角度或者更像自然界存在蛋白的角度评估突变体,对于那些符合进化规则和更像自然界存在的蛋白的突变模型会给出更高的打分。我们在公共数据库中的25个代表不同蛋白质的单点突变数据集[36]上做了测试,使用的无监督模型除上述三种外还有具有代表性的ProGen2[37]和Tranception[38],结果显示25个数据集中,无监督模型在其中15个数据集上的预测结果与实验值的斯皮尔曼相关系数(Spearman correlation)超过了0.5(表1)。无监督模型没有经过额外训练,却可以在代表不同功能指标的多个数据集中取得较好的结果,是因为其学习的内容是自然界中有关蛋白质构成的基础规律,这些规律在一定程度上是和蛋白质活性、热稳定性等具体性质呈正相关的。酶工程中在选择单点位突变位点时,高精度无监督模型的结果对于提高阳性率有重要参考意义。Alper团队[39]利用基于结构的卷积神经网络MutCompute得到10个PET水解酶的单点突变,实验证明其中有8个突变是能提高酶功能的有益突变。经过筛选和组合,该团队得到了能在1周内完全降解大多数PET制品的五位点突变体[39]。在人工智能的辅助下,酶工程已经实现相比于定向进化成本更小、实验周期更短的改造策略。
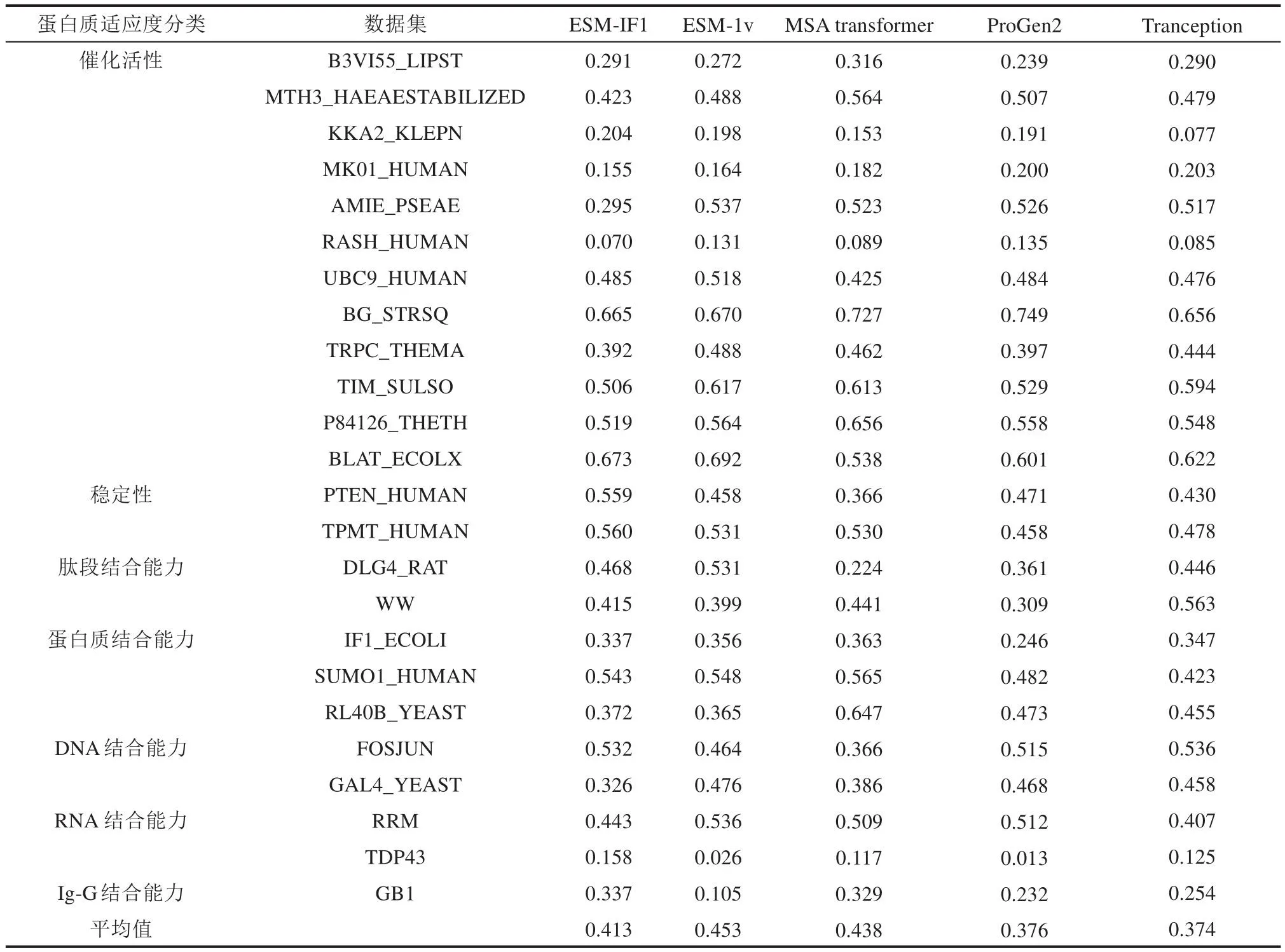
表1 无监督模型在不同数据集上预测结果与实验结果的相关性Table 1 Spearman correlation for predicted fitness developed with unsupervised models
相比无监督学习,监督学习(supervised learning)通过学习某个特定蛋白的突变数据(序列和性质的对应关系)可以更加准确地预测该蛋白突变体的性质。无监督模型已经通过训练学习到了蛋白质的编码方式,因此有监督模型引入无监督模型作为编码模块可以在准确预测特定蛋白质突变体性质的同时保证预测结果符合自然规律。ESM-1b模型使用34层的Transformer在UR50/S数据库上进行预训练,然后使用特定蛋白的突变数据对模型进行微调(fine-tune),得到了相比以往方法更高的精度[40]。ECNet是利用进化环境预测特定蛋白质的突变效果的有监督模型,该模型使用无监督模型TAPE编码蛋白质序列特征,并且从MSA中学习了残基之间的进化约束。ECNet在多个数据集上表现出高于TAPE模型的预测精度,证明MSA中包含的进化信息对预测蛋白质突变效果有正向作用[41]。SESNet是整合了蛋白质序列、MSA和结构信息的有监督模型,在多个数据集上的预测精度超过了现有的监督学习模型[42]。
监督模型往往经过在上千突变体的实验数据上训练后才能达到可靠的精度,但这些数据会造成巨大的实验负担。相比之下,无监督模型在完成繁重的预训练任务后不再需要额外的数据,但是预测精度大多远低于有监督模型。SESNet使用大量无监督模型的预测结果对模型进行预训练(pre-train),之后模型在经过数十条实验数据微调后即可以较高的精度对高点位突变的效果进行预测。测试发现没有经过预训练过程的监督学习无法在使用如此少量的数据进行训练后达到同样高的精度[42]。这套数据增强策略降低了使用有监督模型的实验数据量需求,对指导酶改造更具实践意义。
2.3 蛋白质的从头设计
定向进化的做法是对自然界中已经存在的天然蛋白质进行人工突变改造,使其满足人类工业生产的需求。而蛋白质的从头设计是更加激进且前沿的方法,其目的是创造出自然界中不存在的蛋白质(或者是没有被人类所发现的)以完成人们所需的生物功能。这类研究的代表是华盛顿大学的David Baker课题组,以及中国科技大学的刘海燕课题组。David Baker课题组设计和不断完善的Rosetta软件包,在多个蛋白质稳定性定向进化的案例中取得成功,如中国科学院微生物研究所的吴边团队[17-18,43]筛选设计了耐高温的PET降解酶,以及可催化其他底物的天冬氨酸酶。Rosetta的核心是一整套基于物理参数的分子势能和统计势能的分子力场,其包含了结构生物学中经常提到的氢键、盐桥、溶液可及面积、亲疏水性等作用项。早期的蛋白质从头设计的主要目标是给定模板结构,设计出能够折叠出目标结构的序列[44]。早在2017年,Baker课题组利用 Rosetta设计、Oligo DNA合成技术以及酵母展示高通量筛选技术,完成了多个不同结构域的蛋白质序列设计。刘海燕课题组[45]利用神经网络力场和随机动力学抽样设计出了SCUBA,在此基础上也完成了一系列给定结构的蛋白质序列设计,并经过了晶体结构的湿实验验证。在之后的几年,随着人工智能技术的飞速发展,蛋白质从头设计(结构到序列的映射)问题成为了许多蛋白质结构深度学习模型的基准测试任务,并以此涌现出一系列的AI模型,能更加快速且准确地完成基于蛋白质骨架结构的序列设计,如GVP-GNN、ESM-IF1、ProteinMPNN等。这些模型设计出来的序列往往是能够折叠成所需的蛋白质结构,不过绝大多数都不具有生物催化功能。目前,完全基于结构且设计出具有生物催化功能的蛋白质成功案例很少,如2018年Baker课题组[46]设计了一个β桶状蛋白质,以期实现GFP的发光功能,但是最终设计出来的蛋白质,还是需要人为把发色团分子放进桶状结构内才能实现发光功能,且发光强度比野生GFP更弱。一个里程碑式的研究工作是Baker课题组[47]于2023年设计出的一个荧光素酶:他们开发了一种基于深度学习的方法(family-wide hallucination)以生成大量包含不同形状口袋的蛋白结构,利用这一方法设计人工荧光素酶可选择性地催化合成荧光素底物二苯基特拉嗪(DTZ)和2-脱氧腔肠素(h-CTZ)的氧化化学发光。其中一种小的(13.9 kDa)和热稳定的酶对DTZ的催化效率几乎等同于天然荧光素酶,但底物特异性却高出许多。整体来说,基于结构的蛋白质从头设计方法更加具有创新性和新颖性,但同时成功率也更低,一般需要在万这个数量级的设计序列库上做筛选才能找到阳性序列。这需要有针对性的高通量筛选实验方法,且同时受限于Oligo合成技术的限制,不能在更长的蛋白质序列上做设计合成(一般不超过200个氨基酸)。
另一种蛋白质从头设计是基于特定蛋白质家族的序列设计。其基本的思想是,设计出来的蛋白质序列要尽可能地符合目标蛋白质家族的序列特征。如2020年William P.Russ等[48]利用统计物理中的波特模型(Potts model)学习多序列比对(MSA)中不同位点的氨基酸共进化信息,结合蒙特卡洛采样生成了一批具有催化功能的分支酸异构酶。 目前在人工智能领域,功能强大的生成式模型,也已经被用来辅助生成具有特定催化功能的蛋白质序列以减少后续突变筛选实验的序列候选量。2021年Donatas Repecka等[49]利用GAN生成对抗神经网络,构建了ProteinGAN,设计并筛选出了最高100多个位点突变且具有与野生蛋白质相似催化功能的苹果酸脱氢酶,所有设计的序列中大概24%的序列溶解性良好且具有生物催化活性。最新的结合生成式语言模型的工作中,Salesforce Research的研究人员利用条件生成模型Progen,设计并筛选出了具有生物活性且同已知数据库中的任何蛋白质序列相似度低于30%的lysozyme 序列[50],而且其设计出来的序列阳性率很高,能够达到60%。受益于目前人工智能自然语言处理领域的发展,蛋白质序列可以被看成是一种蛋白质语言,这项工作为蛋白质从头设计提供了新的思路。蛋白质语言模型首先在公共蛋白质序列数据库上进行预训练,模型学习到了蛋白质序列中氨基酸的排列规则(类似于蛋白质的一种语言规则),之后其可以对任何序列是否接近自然序列做出判断。一般来说,更符合自然序列特征的序列,意味着其具有更好的结构折叠能力和更好的表达能力以及水溶性。在需要对特定功能的蛋白质做设计之前,将预训练模型在这些特定家族的蛋白质序列上进行微调(finetune),然后其对特定功能的蛋白质序列具有更准确的生成和预测能力。人工智能领域大热的diffusion扩散生成模型,在蛋白质设计上的应用还主要集中在结构到序列的生成任务上,如Baker组做的ProteinMPNN,目前还没有公开的利用diffusion生成模型设计出具有生物催化功能的蛋白质序列。diffusion模型相对于传统的GAN具有更强的生成能力且更容易训练,不过diffusion模型本身更适合在连续空间生成,如图像音频数据等;而蛋白质序列生成是个典型的离散空间生成问题,其每个位点只有20种可能,相对而言自然语言处理中的GPT式的生成模型更适合蛋白质序列生成。GPT类的生成模型天然地可以用到蛋白质序列生成任务上来,2022年Noelia Ferruz等将开源的GPT2.0模型框架在蛋白质序列数据库上做了训练,得到了ProtGPT2,作者用AlphaFold2对生成的蛋白质序列进行折叠发现,其生成的序列在二级结构上与天然蛋白质相似,且ProtGPT2还生成了自然界中不存在的蛋白质结构,不过这些结果还有待湿实验的进一步验证。
2.4 人工智能技术与采样方法
使用计算机代替高通量筛选方法去探索庞大的序列空间可以大幅度缩小实验成本,相比定向进化方法,高精度的模型可以更快地找到最优突变体,从而减少实验周期。但是蛋白质多点位突变的序列空间非常庞大,即便使用计算方法也无法完全遍历,因此需要按照一定方法对序列空间进行采样。传统采样方法包括随机突变、贪婪算法和蒙特卡洛模拟退火等[51-52]。其中随机突变方法即在序列空间中随机采样,采样结果将被计算方法筛选。这种采样方法效率较低,并且找到最优突变的概率严重依赖采样数量。贪婪算法先选择一批表现较好的突变体作为亲本(parent sequences),然后迭代组合这些突变生成子本(children sequences)。该方法可以有效探索高维突变的序列空间,但是探索内容受到亲本限制,无法在整个蛋白质的序列空间中进行有效检索。蒙特卡洛方法即在一个不具有物理意义的玻尔兹曼分布中采样。该分布的定义为pi=(1/Z)exp (-yi/kT),其中yi是计算方法对序列的预测结果,k为常数,T为温度,Z是归一化系数。除以上方法外,Hu等[53]利用贝叶斯优化方法指导定向进化,将采样过程和代理模型迭代优化过程结合起来,经过4轮迭代,成功在RhlA酶的四点突变序列空间中找到能使产物选择性提升4.8倍的突变体。Krishnaswamy团队[54]使用深度学习方法对蛋白质进行编码,然后在正则化隐空间中使用梯度上升的方法寻找极大值点,被还原到序列空间的采样结果将被认为是有益突变,但这种方法的有效性还有待在湿实验层面验证。在计算机都无法遍历序列空间的情况下,采样方法直接影响计算机找到最优突变体的概率。高效、可靠的采样方法可以让计算机辅助的酶工程更容易找到符合预期的序列。
3 结论与展望
经过多年的发展,在酶工程的定向进化、理性设计、半理性设计和人工智能辅助设计等不同方面都有重要工作涌现。理性设计建立在研究者对结构以及催化机理深入了解的基础上,可以改造酶的选择性,构建野生型不存在的新反应。定向进化突破了酶工程在酶催化机理、结构和具体氨基酸替换的影响等方面的知识壁垒,让研究者不需要了解蛋白质也可以进行改造,但定向进化方案需要面临筛选成本过大和实验周期过长的问题。半理性设计将序列空间限定在一个更小也更合理的范围内,减少了筛选成本。在合适的采样方法的引导下,深度神经网络模型可以在酶的活性、热稳定性甚至是选择性等功能的改造上给出置信度较高的建议,但作为一种数据驱动的计算方法,其预测结果受到训练集和采样策略的限制。这些方法虽然原理不同,但可以在具体案例中被结合起来。专家在半理性设计中提取的特征可以作为机器学习方法的输入,人工智能建议的阳性突变也能被用作定向进化的起始位点,这些组合都有成功案例。
目前人工智能辅助酶工程领域正处于飞速发展阶段。各种神经网络模型正在向更准确、更高效的方向快速更新迭代。神经网络的学习对象从早期的手工特征逐步变化到以蛋白质序列、MSA和结构为主的原生信息,代表着人工智能学习高维信息的优势正在被逐渐放大。为了应对大规模基因测序带来的蛋白质序列数据库的爆炸式增长,目前最大的蛋白质语言模型的参数量已经达到了150亿[55],这类突破是生物、计算机和人工智能等多个学科交叉深化的结果。目前酶工程领域的人工智能方法仍然需要在预测精度和学习蛋白质上位性等方面做出突破,具备高泛化能力和快速采样能力的高性能神经网络模型将是生化实验室降低定向进化成本与实验周期的关键工具。酶工程的数字化计算设计已经成为未来的趋势。
