基于靶标结构的环肽分子计算设计
2023-07-10王凡灏来鲁华1张长胜1
王凡灏,来鲁华1,,3,张长胜1,
(1 北京大学前沿交叉学科研究院定量生物学中心,北京 100871; 2 北京大学化学与分子工程学院,北京分子科学国家研究中心,北京 100871; 3 北京大学-清华大学生命科学联合中心,北京 100871)

1 环肽药物
蛋白质-蛋白质相互作用在生物体系中发挥了关键作用,很多相互作用体系,如酶/抑制剂、细胞因子/细胞膜受体、病原体/受体、细胞信号转导蛋白质作用、分子机器组装体内部作用、蛋白质聚集等都被认为是潜在的重要靶标[1-4]。通过调节蛋白质相互作用可以提高药物的专一性,减少药物的毒副作用。蛋白质-蛋白质相互作用已经成为国内外药物研发机构竞相投入研发的药物靶点[5-10]。
蛋白质-蛋白质相互作用界面一般较大而平坦,较难进行高专一性结合的小分子化合物设计。蛋白质-蛋白质相互作用界面面积通常为1500~3000 Å2,而小分子化合物总的溶剂可及表面积只有约150~500 Å2。相对于小分子,多肽分子可以提供较大的结合界面,并且具有易合成[11]、结合专一性强、毒性小、免疫原性小等独特的优势[12-16]。使用天然多肽作为药物,在调控分泌蛋白、细胞膜上受体等蛋白质功能方面已取得了很大成功[17]。但由于天然多肽在很多情况下存在热稳定性差、易被蛋白酶降解、结合较弱的缺点,导致这些多肽分子在体内的半衰期短、药效弱[18-21]。所以,改造这些多肽分子或全新设计稳定的多肽是药物研发的重要目标[22-23]。
多肽分子的环化,如主链首尾形成酰胺键、残基侧链间形成二硫键或其他共价键,可以限制多肽的构象,是提高多肽药物稳定性的重要手段。环肽降低了肽链的柔性,因此还可以减少与靶蛋白结合的熵损失[24-30]。在天然氨基酸环肽的基础上再引入N-甲基化氨基酸、D型氨基酸、类肽等非天然残基可以更近一步解决易被蛋白酶水解等问题,并进一步拓宽其可设计化学空间[31]。天然产物与人工设计的环肽分子越来越多地用作调节蛋白质-蛋白质相互作用,例如HDAC抑制剂[32-34]、灰霉素[35]、CXCR4拮抗剂[36-37]、抑制HIF-1a/HIF-1b的cyclo-(CLLFVY)[35,38]等。
过去20年里经FDA批准上市的环肽药物有18种[39-40],包括激素类药物和靶向药,表1列出了这些药物的结构和功能。
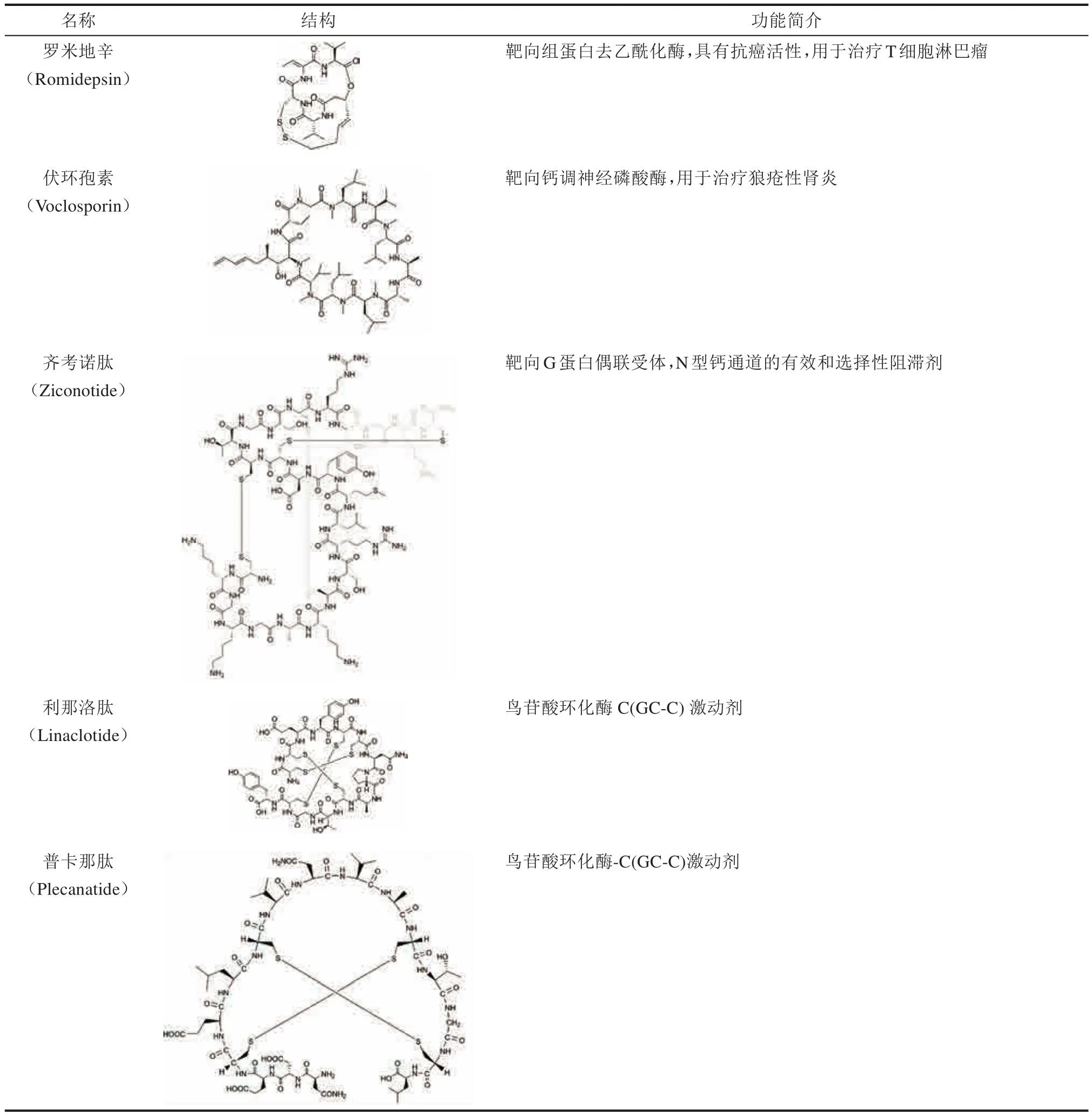
表1 已获FDA批准的18种环肽类药物汇总表Table 1 Summary of 18 cyclic peptide drugs approved by FDA
这些分子主要来源于从自然界中分离的环肽,经改造优化提高了效价、药代动力学和药代动力学特性。例如通过将芳香萘和D-色氨酸加入生长抑素类药物兰瑞肽(Lanreotide,结构见表1),可以稳定其超分子堆积的纳米管,有助于延长其半衰期、抑制激素水平和活性的能力。从技术角度来看,环肽药物的开发得益于快速发展的化学合成与修饰方法和现代分子生物学技术,环肽的分子结构不仅可以进行灵活修饰,一级序列也可以快速突变和优化,以获得更高的产量与生物活性。例如由环孢素衍生而来的伏环孢素(Voclosporin,结构见表1)就是利用化学修饰,将E-MePmt1分子基团取代环孢素A(CsA)中第一个残基Bmt1[41-42],以优化其对靶标的结合能力,并提升了代谢稳定性[43-45]。但是单纯通过大规模实验分离与筛选的方法获得可结合靶标蛋白环肽分子的效率低、成本高,所以计算设计方法将极大地帮助环肽药物的开发[46-48]。
2 环肽的结构与环肽-靶标蛋白复合物结构
环肽的三维结构和环肽与靶标蛋白的复合物结构为理解环肽性质和功能机制提供了重要依据。已有很多工作收集了自由环肽的实验结构,例如,Beaufays等[49]的研究中收集了38个PDB数据库中的环肽结构作为测试集,长度5~30个残基,通过侧链或主链之间的共价键连接成环;黄胜友实验室[50]在多肽构象生成方法MODPEP2.0的研究中,收集了蛋白质结构数据库(PDB)中采取二硫键连接成环的环肽结构。我们对PDB中的环肽配体与靶标蛋白的高分辨(≤2.5 Å)复合物结构进行了统计分析,截至2022年9月,PDB中包含了经过去冗余的88个环肽配体与靶标蛋白的高分辨复合物结构,这些结构中的环肽配体长度为5~21个残基,靶标蛋白在相互作用界面上被埋藏的面积大部分在300~700 Å2之间(见表2)。

表2 PDB数据库中的环肽-靶标蛋白质复合物结构数据表Table 2 Non-redundant cyclic peptide-target protein complex structures in the PDB database
这些结构中,31个环肽配体通过酰胺键环化,23个环肽配体通过二硫键环化,41个环肽配体中含D型氨基酸或其他非天然氨基酸,或者是引入了新的基团实现合环,例如图1(c)的环肽是由线性肽两末端的半胱氨酸通过与α,α'-二氯-间二甲苯反应形成的合环[51]。其中不含D型氨基酸环肽的主链二面角φ和ψ分布与天然蛋白的Ramachandran统计分布相近,如[图1(a)]所示,这些二面角大都在Ramachandran统计允许的范围之内。Riniker团队[52]对PDB数据库和CSD数据库中的大环化合物(包括环肽)的统计结果表明,环上可旋转单键数≤8时,才会导致环上的二面角分布受环应力的影响而偏离相应线性分子的分布范围[52],而环肽分子的环内一般含有≥5个残基,可旋转单键数一般≥10。含D型氨基酸的L/D混合环肽的主链二面角ψ和φ的分布[见图1(b)]趋近于甘氨酸的非手性Ramachandran统计分布。

图1 PDB中环肽-靶标复合物数据集(见表2)中环肽配体的参数统计图(a)、(b)中蓝色背景分布为天然蛋白质体系的氨基酸残基扭转角ψ/φ的分布。(a)仅含天然氨基酸残基的环肽配体主链扭转角分布图(ψ/φ);(b)存在非天然氨基酸残基的环肽配体主链扭转角分布图(ψ/φ);(c)数据集中所有环肽配体环序列长度分布图;(d)数据集中所有环肽配体与靶标之间界面面积分布图Fig.1 Parameters of cyclic peptide ligands with the cyclic peptide-target complex data set (see Table 1) in PDB Distribution of torsion angles of the main chain of cyclic peptide ligands containing natural (a) and non-natural (b) amino acid residues (ψ/φ), in which blue cloud highlights the distribution of the torsion angle ψ/φ of amino acid residues in natural proteins; Length distribution of the loop sequences of all cyclic peptide ligands in the data set (c); Distribution of the interface area between all cyclic peptide ligands and targets (d)
此外,有些环肽结构中存在一些较为刚性的局部结构以稳定分子骨架,从而减少与靶标蛋白结合过程中的熵损失,增强环肽配体与靶标蛋白的结合[26-27,30,53-54],如图2中的β发卡末端环化形成的环肽配体结构[(a)以侧链二硫键合环,(b)以首尾酰胺键合环],β片层之间的氢键和β转角处形成的1,3、1,4氢键相互作用,稳定了环肽的构象,增强了骨架的刚性。脯氨酸也可以限制环肽主链的运动,因此在刚性环肽骨架的从头设计工作中引入脯氨酸也是稳定目标骨架结构的重要策略[55-56]。

图2 PDB编号为5DJC(a)、4K1E(b)和5NES(c)的复合物结构环肽配体结构示意图Fig.2 Cyclic peptide ligand structures for the complexes in PDB 5DJC (a), 4K1E (b) and 5NES (c)
分子动力学模拟(MD simulation)是研究蛋白质和多肽结构动态变化的重要手段。基于分子动力学模拟的环肽药物分子的构象研究推动了研究者们对环肽构象的深入理解与经验积累,为实现大规模环肽分子从头设计提供了理论依据[57]。由于蛋白质中的环结构(loop)与环肽结构较为相似,通用的分子动力学模拟力场参数根据蛋白质loop结构进行重新拟合调整后可以更好地用于环肽分子的结构优化与构象采样[58-63]。Geng等[64]就曾以晶体结构为基准,比较了四种多肽力场Amber99 sb-ildn[65]、OPLS-AA/L、RSFF1[66]和RSFF2[67]预测全反式(酰胺键)环肽结构的能力。其中基于蛋白质环形结构(loop)库进行参数化的RSFF1和RSFF2力场得到的预测结果与晶体结构匹配最佳[68]。同其他柔性体系一样,增强采样的方法可以加速环肽构象采样的过程,提升采样的效率。其中最为广泛使用的是副本交换分子动力学(REMD)[69-71]和元动力学(META)[72-73]。
3 环肽分子结构建模(结构预测/构象生成)
环肽分子结构建模的目标是生成环肽的低能量的合理构象,对于较刚性的环肽,如环化的β发夹,就是预测出准确的折叠结构。从结构数据库中收集环肽结构建立数据集来检验由环肽结构建模算法的表现。计算生成的环肽构象是环肽与靶标蛋白对接筛选的前提。经检验,环肽分子的结构采样方法以及打分评价方法,可直接用于大规模的基于靶标结构的环肽分子的改造、生成、评价[74]。
3.1 基于模板结构的比较模建算法
黄胜友实验室[50]开发的MODPEP2.0(http://huanglab.phys.hust.edu.cn/software/modpep2)可以快速地产生二硫键环化的环肽构象。他们从PDB数据库中收集了3~30个氨基酸长度的二硫键环肽结构,通过聚类选择后得到的不同长度的二硫键环肽结构集合作为目标环肽构象生成的模板库。MODPEP2.0选取与目标肽序列相似性高的,并且结构分辨率高的结构做模板,选取概率如下:
式中,si是基于目标序列和第i个结构的序列相似性;ri是第i个结构的分辨率,除以相应的最大值(smax,rmax)是为了对不同的打分项进行归一化;w为两项的权重系数。之后在选定的模板主链上应用侧链构象转子库逐个残基安装侧链,得到目标二硫键环肽的构象,并通过选取不同的模板生成多样的构象[50]。对于环以外序列的结构,算法用MODPEP的早期版本通过片段组装方法完成[75]。该团队构建了测试方法对MODPEP2.0生成构象的准确性进行评价,当生成10种或100种构象时,与测试数据集中结构最接近的Cα RMSD平均值分别为2.20 Å、1.66 Å[50,75]。
张阳实验室[76]发展的I-TASSER是基于结构采样和优化作蛋白质结构预测的代表性方法之一,加入环化所需的共价距离约束就可用于环肽结构预测[76],提供在线计算服务(http://zhang.bioinformatics.ku.edu/I-TASSER)。该算法通过多种序列比对方法识别与连续片段相似的PDB模板结构,将模板的主链作为这个片段的主链结构,并通过基于模板片段副本交换的蒙特卡洛采样方法探索整个蛋白质或多肽的结构空间,获得能量较低的结构模型。环肽中的环化共价键可作为几何约束加入能量函数。在Peplook的环肽结构测试集中,I-TASSER表现优异[49,76]。
3.2 基于主链结构预测和分子力场优化的方法
PEPstrMOD算法(https://webs.iiitd.edu.in/raghava/pepstrmod/)从序列预测所得二级结构(α螺旋、β片、转角、无规等)类型构建多肽的初始主链构象,使用Amber力场对结构进行能量最小化和动力学模拟优化[77]。对于环肽,在分子拓扑结构中加入相关共价键连。PEPstrMOD将末端修饰、D-氨基酸、非天然氨基酸、翻译后修饰等残基类型的力场参入加入Amber力场,使之可以预测含有非天然氨基酸残基的多肽结构[77-79]。PEPstrMOD算法在来自于PDB数据库的ModPep、ModPep16、CyclicPep三个数据集上进行了检验,预测得到的主链原子的均方根偏差为3.81~4.05 Å之间。
PEP-FOLD算法(https://mobyle.rpbs.univ-parisdiderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3)也是先从序列预测各残基处的主链构象,然后利用力场做优化。不同的是PEP-FOLD将主链二级结构扩展为27种类型,基于预测的所有位置的这27种类型的概率分布,使用贪婪算法构建粗粒化的三维模型,使用蒙特卡洛方法作粗粒化力场优化后再构建全原子模型。同样地,在分子拓扑结构中加入相关共价键连用于环肽的结构预测[80-81]。PEPFOLD的最新版本改进了粗粒化力场的范德华项和用于主链结构预测的片段库,提高了多肽结构的预测性能[80-83]。
3.3 基于主链扭转角采样的环肽结构预测算法
环肽局部连续片段的构象运动是两端都受约束的运动,与力学中的运动学闭环(kinematic closure,KIC)类似,例如对于机器人,在给定肩部和指尖固定位置的情况下,确定机器人手臂内部关节的可能位置与扭转角度范围。Rosetta(https://www.rosettacommons.org/software)中的genKIC是运动学闭环算法在蛋白质环区结构预测、蛋白质主链结构扰动中的成功应用。genKIC算法选择一肽段上第一个、中间某一个和最后一个残基的Cα原子固定为转动中心,然后对非转动中心的残基用Ramachandran概率对ψ/φ随机采样并对N-Cα-C键角采样,得到的肽链在固定残基处断开,之后运动学闭环算法找到使肽链重新闭合的3个转动中心残基的ψ/φ值,从而获得肽段的新构象[84]。使用Rosetta势能函数和蒙特卡洛采样实现对构象空间的探索,获得蛋白质环区或环肽的低能构象[85-86]。
Peplook(http://www.biosiris.com/en/Online_order/PepLook/PepLook_order.html)是一种对蛋白质或多肽的主链二面角进行玻尔兹曼随机采样来预测结构的算法[49,87]。Peplook从64对ψ/φ角度中对每一个氨基酸随机取值[88-90],根据这些主链二面角值构建结构,并计算体系能量,在环肽结构预测中体系能量加入首尾二硫键或酰胺键原子的距离相关项。每一轮生成1万个结构,前一轮得到的体系能量和主链二面角取值的关系决定下一轮中各ψ/φ角度对被取到的概率,采样100~500轮得到低能量构象[49]。在38个环肽测试集上,Peplook建模最佳结构的主链原子的均方根偏差的平均值为3.8 Å[49,91]。
3.4 基于距离几何的构象生成算法
构象生成器ETKDG是小分子化合物构象生成最常用的方法之一。ETKDG通过分子的拓扑结构和晶体结构中的二面角数据,生成分子的距离边界矩阵,根据该矩阵限定的原子间距离的范围随机产生一个距离矩阵,然后由距离矩阵产生三维结构,之后优化构象[92]。最近该方法改进了对大环化合物分子和环肽的生成。mETKDG使用椭圆几何约束来限制环肽整体环系骨架,并且加入可调整的库仑相互作用作为酰胺原子之间的奖励式方法来模拟跨环分子内氢键[52]。mETKDG的环肽建模性能在由PDB、CSD 筛选的环肽分子测试集上得到了验证,mETKDG的重构主链原子的均方根偏差的平均值为1.23 Å。目前mETKDG已经被写入常用的RDkit(http://www.rdkit.org/)Python工具包中[52,92]。
4 基于靶标结构的环肽分子计算设计
基于靶标结构可以改造设计或全新设计与之结合的环肽分子,在具有备选的环肽库的情况下,通过基于分子对接的虚拟筛选,可以缩小实验测试的分子范围,从而更高效地发现新的结合靶标的环肽分子。分子动力学模拟是详细研究和改进已知环肽分子的重要方法。近年来,以Rosetta为代表的从头生成算法大大拓展了设计环肽分子的化学空间,为针对特定靶标结构设计全新的环肽药物分子提供了解决方案。对于细胞内的靶标蛋白,需要在设计中考虑跨膜活性。因此本节将介绍这4个方面的计算设计方法(图3),表3总结了这些算法的主要思路和适用目的。
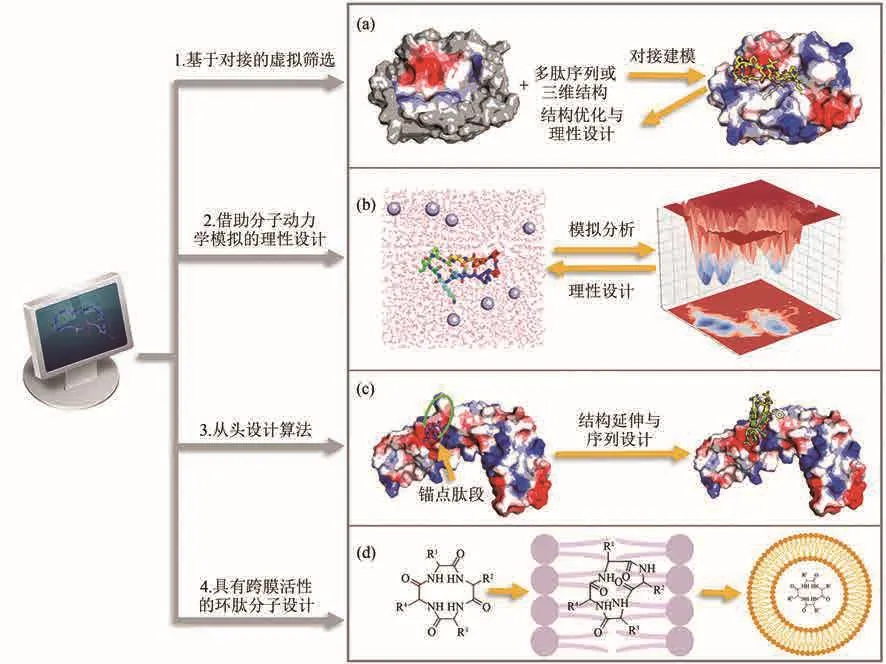
图3 基于靶标结构的环肽设计算法(a)基于分子对接的虚拟筛选算法;(b)基于分子动力学模拟的理性设计算法;(c)从头设计算法;(d)跨膜环肽分子的设计算法Fig.3 Overview of computational methods for target structure based cyclic peptide design(a) Virtual screening algorithms based on molecular docking; (b) Rational design algorithms based on molecular dynamics simulation;(c) De novo design algorithms;(d) Design algorithms for transmembrane cyclic peptides

表3 基于靶标结构的环肽分子计算设计算法Table 3 Structure based computational design algorithms of cyclic peptides
4.1 基于分子对接的虚拟筛选方法
基于大规模环肽数据库的虚拟筛选是最早出现的计算设计方法,环肽结构库和对接及打分算法是这一方法的核心内容。
Duffy等[95]于2011年便尝试过生成用于虚拟筛选的环肽数据库,开发了CycloPs,该程序可以根据规则与用户预定义的约束,大量生成一维SMILES,通过RDkit建模为三维环肽分子结构,结合后续的虚拟筛选构成了完整的环肽药物的设计流程。但是环肽药物发现的虚拟筛选方法往往局限于数据库的规模与复合物对接建模的准确度。
Sanner实验室[96-97]开发的AutoDock CrankPep(或简称ADCP)是由小分子-蛋白对接软件AutoDock改进而来的多肽(环肽)对接软件(https://ccsb.scripps.edu/adcp),可以对接长度在20个氨基酸残基以内的线性多肽与环肽结构。他们应用了基于多肽骨架的曲轴运动微扰方式,支持对具有环肽主链的构象进行高效采样,并在AutoDock基本的Monte Carlo采样过程中引入了主链环化势能项,使得程序能够从线性多肽序列出发的扩展线性肽开始对接计算,从而在没有初始环状构象可用的情况下实现对接与环化同时进行,最终得到环肽-蛋白复合物结构。最后,距离依赖性势能确保多肽环化。这种势能不同于之前的基于环化势能的方法,ADCP中线性多肽能够在模拟过程中环化、断裂和重组[96-97],中间状态的存在允许算法在对接过程中探索主链的各种构象组合并确定最佳二硫键或首尾环化位点。ADCP测试的平均fnc(对接得到的复合物构象对于天然复合物构象中的原位配体-受体相互作用对的召回率,在蛋白质复合物结构预测比赛CAPRI中,fnc高于0.3对应中等对接精度,fnc超过0.5则认为对接精度较高)超过0.5[98]。ADCP是环肽-受体复合物相互作用建模和结构预测的有效工具,可用于开发设计环肽药物,并在相当一段时间内被认为是该领域最先进的SOTA(state of the arts)算法[93]。
与ADCP类似,HADDOCK算法的最新2.4版本(https://wenmr.science.uu.nl/haddock2.4)在对接过程中也应用了末端二硫键几何约束或者首尾氨基酸距离约束,使多肽结构在对接过程中逐步环化[94]。在对接流程中,用于对接的环肽是用PyMOL的内置功能从多肽序列生成起始构象[99],HADDOCK会利用环化约束指导线性多肽结构逐步收紧多肽首尾或二硫键之间的距离(通过HADDOCK)使线性肽强制环化。环化之后,HADDOCK会将生成的环肽结构分别与各自的受体结构进行对接,并实施9种对接方案以获得高质量的配体结构[94]。HADDOCK在环肽-蛋白质复合物建模方面的表现与ADCP基本相当[96][在完全未结合状态从头对接的情况下与ADCP的表现相当,在重建模(redock)性能评估时略优于ADCP(HADDOCK中的默认模式)]。由于HADDOCK还可以结合各种实验数据(例如关于肽构象的核磁共振谱图信息)来指导对接,因此基于已知结构的测试性能可被认为是下限,通过额外的实验数据输入可以进一步增强算法性能[94]。环肽配体的对接建模为环肽药物分子的虚拟筛选与理性设计提供了高效可靠的工具。
4.2 借助于动力学模拟的设计方法
较为传统的药物分子改性与理性设计方法仍然适用于设计环肽配体,这类方法向环肽复合物体系的迁移应用往往依赖于高效的环肽配体分子构象采样算法。
Razavi等[79]使用计算筛选和REMD模拟相结合的算法,通过模拟LapD中的β-发夹(hairpin)结构,成功设计了环肽配体。LapD是一种细菌蛋白,其与LapG的相互作用对生物膜的形成至关重要[78-79,100-101]。该团队首先使用隐式溶剂模型的REMD 模拟用于快速筛选潜在的环肽模拟物[69],随后应用显式溶剂模型进一步进行模拟采样结合虚拟筛选方法得到4种最有利的设计产物,最终成功设计出了类似LapD β-发夹结构的一系列环肽配体[73]。若已知体系的决速自由度,则可以应用BE-META模拟方法以实现高效构象采样。例如,在基于isoDGR的αvβ3环肽拮抗剂的设计中便使用了BE-META结合多构象对接的方法[102],从而在其构象系综中发现最有利于结合的配体构象,并根据这些优势构象进一步修改isoDGR分子结构,衍生物的平衡系综更倾向于与受体产生强结合的状态[69]。
4.3 从头生成设计方法
从头设计方法可以得到与已知肽段结构完全不同的全新结构环肽,需要同时考虑生成环肽结构的稳定性以及跟靶标蛋白的亲和力,在环肽结构采样的同时优化与靶标蛋白的计算结合能。
在不考虑靶标的自由环肽结构生成方面,Hosseinzadeh等[55]利用基于Rosetta软件的genKIC算法生成L/D氨基酸混合的7~14个残基的环肽的主链骨架结构。用全甘氨酸的序列和甘氨酸、脯氨酸非手性的Ramachandran概率对ψ/φ随机采样,生成闭环的主链骨架,最终得到200多个预测可折叠成单个稳定结构的环肽,其中12个结构得到实验验证。此工作的算法流程支持加入D-氨基酸来拓宽构象空间并且能够形成双环(同时存在首尾酰胺键连接与二硫键连接)拓扑结构以增强大环刚性,这些结构几乎完全覆盖了已知环肽化合物可能的构象空间,并探索到了大量非天然的局部二级结构,大大拓宽了理性环肽药物设计和虚拟筛选方法的可用起始结构空间[55]。之后,Hosseinzadeh等[34]在2021年提出了锚扩展(anchor extension)方法,将genKIC环肽结构设计方法用于直接在靶标蛋白表面设计环肽配体,是一次里程碑式的尝试。这种方法需要目标受体表面的三维结构,并利用已知有较强结合配体分子的对结合能贡献较大的基团作为锚,置于原位(同时删除配体的其余部分结构),锚即为环肽结构向外延伸的起始位点。锚扩展方法使用Rosetta软件中的广义运动学闭环方法(genKIC)在锚点周围构建环肽骨架,生成的合理大环骨架从锚点出发向外延伸进行主链优化采样与序列设计,逐步增强其与受体之间的相互作用[84-86]。作者选定的测试体系是HDAC2和HDAC6分别与一种蓝藻毒素Largazole结合的复合物结构,保留其中的2-硫-2-氨基-7-磺酰基庚酸(SHA)的部分作为锚,来从头设计环肽配体[103-104],最终得到了多个IC50为纳摩尔量级的产物(HDAC2与SHA的IC50和与环肽配体最优IC50分别为5.4 μmol/L、9.1 nmol/L,HDAC6则为0.6 μmol/L、5.4 nmol/L)。
锚扩展方法需要结合较强的“锚”残基结构作为起始。Delaunay等[105]提出的Des3PI算法(https://github.com/des3pi/Public_Des3PI)能够生成这些“锚”残基,该团队利用20种氨基酸残基与靶标蛋白表面进行对接筛选,得到结合较强的位点,然后将对接在表面的氨基酸残基间通过甘氨酸简单连接成环得到环肽配体分子[105]。这种方法普适性较强,但是由于主链结构柔性较大,生成的环肽配体刚性与结合稳定性弱于锚扩展工作中精细设计的环肽配体结构。
4.4 具有跨膜活性的环肽分子设计
药物分子的跨膜活性决定了其是否可以作用于细胞内的靶标[39]。多数多肽药物的靶标为细胞膜上的受体,近年来在可以进入细胞内的多肽设计领域取得了重要进展。实验研究表明主链酰胺键的N-甲基化修饰以及降低侧链的极性均可提高环肽的跨膜活性[39]。Sindhikara等[106]通过动力学模拟的方法发现,环肽分子的主链往往在磷脂双分子层内会将极性的羰基氧和氮上的氢原子通过环肽分子的构象变化(酰胺键顺反异构转变)收容到环内,这大大降低了分子的极性,使之与磷脂细胞膜融合,穿过细胞膜后又可以被极性较大的细胞内环境诱导,使环肽分子采取相对较为极性的构象,易于与极性的蛋白靶标结合,形成较为稳定的复合物[18,107-108]。因此此类环肽分子具有随环境极性不同转变构象的特性,这些特性决定了此类环肽具有作为可跨膜药物分子的潜力[109-110]。Baker实验室[56]综合了这些特性进行了具有优良膜渗透性的环肽设计并且取得了成功。该团队改进了genKIC算法,在采样的过程中,允许整个化学基团(如氮甲基)被放置,这就可以在环肽结构中引入非天然氨基酸修饰,并且Rosetta的残基类型被重新整理为残基和补丁,这些补丁可以通过Rosetta的能量函数来对已有的化学实体做化学修饰[111]。研究者们利用优化调整后的Rosetta软件设计了一系列长度为6~12的环肽结构,这些结构均为化学修饰或支持构象转变的刚性环肽结构,随后进行了实验合成与结构解析,实验测出的结构与设计结构叠合较好(BB-RMSD<1.5 Å),最后测试了新结构的膜通透性和口服生物利用效率,得到了较好的效果[56]。此工作在环肽药物设计做了进一步的探索,从化学修饰角度着手,大量设计结构稳定且具有跨膜活性潜在环肽药物分子,为环肽药物分子的虚拟筛选工作进一步提供了可靠基础。
5 总结与展望
目前环肽作为调控蛋白质-蛋白质相互作用的重要研究对象,已经获得了越来越多的关注,有关环肽分子的结构、动态构象、与靶标蛋白质相互作用的实验数据也逐渐增多。基于这些实验数据,研究者通过分子动力学模拟、分子构象采样等方法研究了环肽的构象空间,开发了基于比较模建、基于主链扭转角采样、基于主链二级结构预测和分子力场优化和基于距离几何的环肽分子构象生成或结构预测算法。蛋白质中环区(loop)与环肽的结构和动态性质相似,这些结构数据也是进行环肽构象研究和结构建模方法测试的重要数据来源。以这些研究为基础,人们已经开始探索基于分子对接的虚拟筛选方法、借助于分子动力学模拟的方法和基于结构采样的从头生成方法来针对目标的靶标结构进行环肽分子的计算设计,近些年对环肽类化合物的生物膜透过性的研究也取得了进展。表4总结了这些环肽分子结构建模和基于靶标结构进行设计的方法。基于靶标结构进行环肽设计的关键是能够同时准确高效地评估环肽分子结构构象的稳定性、环肽配体与蛋白质靶标结合强度。更加高效准确的环肽序列和构象采样算法、环肽与靶标蛋白质结合能的评估方法准确,将提高环肽分子计算设计的效率和成功率,从而推动环肽药物与功能化环肽分子的开发。另外,对已有的环肽药物的性质和功能机制进行研究和学习,以及将计算设计方法和噬菌体展示、酵母展示等高通量实验筛选技术相结合,是促进环肽药物开发的途径[112]。
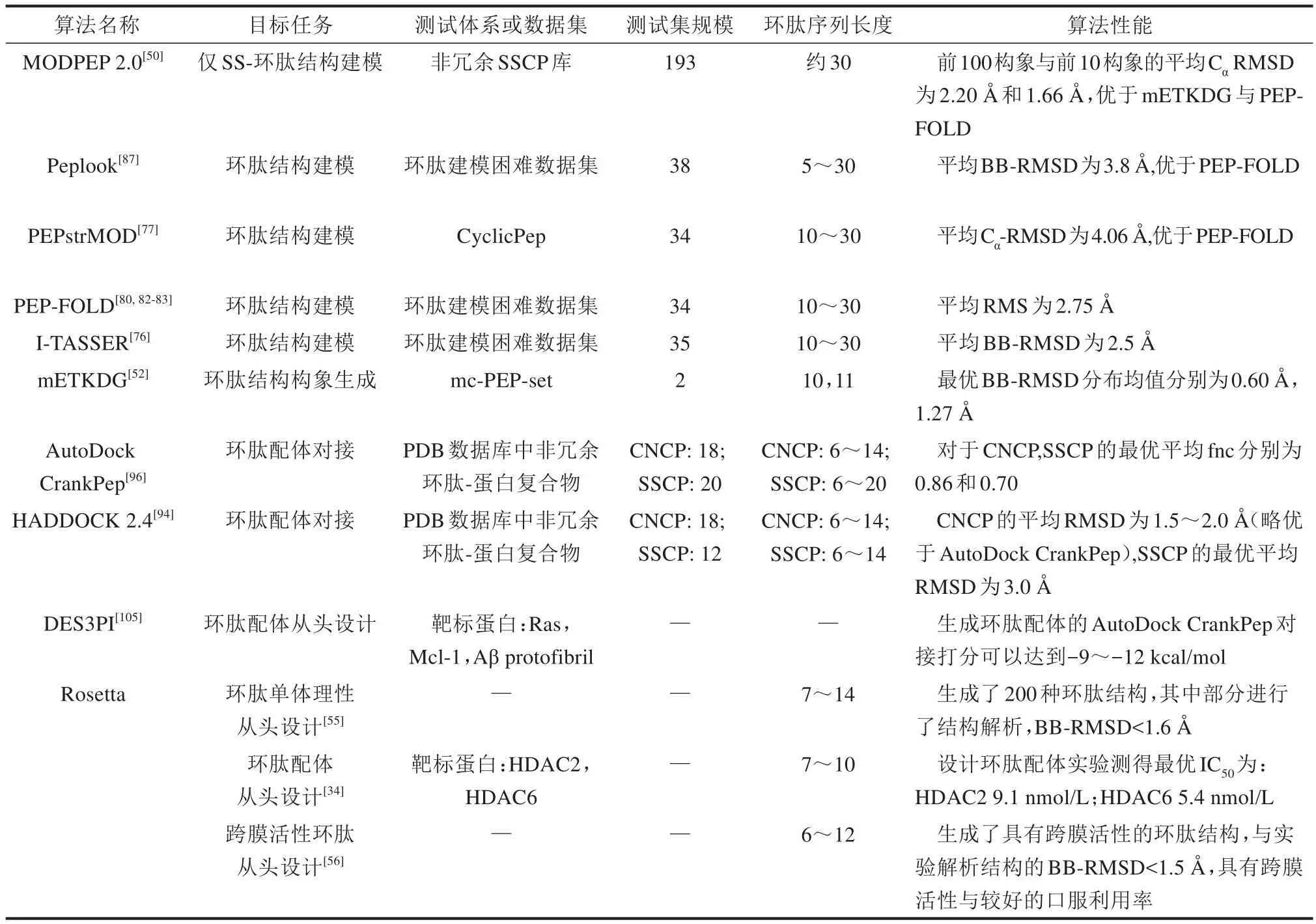
表4 本文所涉及的算法总体简介Table 4 A brief introduction of algorithms included
近年来以深度学习为代表的机器学习技术快速发展,在图像识别、语音识别以及自然语言处理等领域取得突破。在蛋白质结构研究领域,以AlphaFold2为代表的基于深度学习技术的端对端方法展现出令人惊叹的由蛋白质序列预测折叠结构的效率和精度,显著超越了基于经验势能打分和采样的方法。结构设计是结构预测的反问题,机器学习技术正在推动蛋白质或多肽设计从基于物理势能函数的阶段到数据驱动的新阶段。例如目前基于深度扩散模型的RFdiffusion[113]、Chroma[114]等工作,已经实现了多种场景下的蛋白质(多肽)结构从头生成,基于靶标结构的小分子配体也可以通过深度学习模型从头生成,比如LiGANN[115]、DeepLigBuilder[116]等。这些生成模型的成功依赖于大量的已知三维蛋白质与复合物结构数据,基于机器学习的环肽结构生成算法或许可以借鉴蛋白质或多肽设计的相关算法,并通过引入蛋白质环区结构的数据以弥补环肽结构数据少的不足。另外,环肽设计领域未来的挑战是更加多样化的功能环肽的从头设计,首先需要进一步拓宽环肽的可设计化学空间[117-121],比如引入非天然氨基酸、N-甲基化修饰与更丰富的环化方式等等[122-124]。在设计更多可用环肽骨架的同时能够引入非天然的化学修饰改性,并对修饰后的分子进行有效模拟采样与结构优化,实现对环肽分子多样化设计与功能调控。此外,自然界中的环肽分子不只是单环结构,通过提取天然产物或基于较为传统的环肽合成方法即可产生双环肽、三环肽等较为复杂的主链环系拓扑结构,这些额外的环状结构可以进一步提供空间约束以增强分子刚性,但同时也大大增加了构象空间复杂性[125-126]。然而目前对于环肽体系的计算研究大多停留于单环结构。这些需求与挑战为环肽设计领域未来的研究提供了契机。
