数据驱动的酶反应预测与设计
2023-07-10曾涛巫瑞波
曾涛,巫瑞波
(中山大学药学院,广东 广州 510006)

酶是自然界中的能工巧匠,其以高效、精准的手段催化生物体内大量化学反应[1]。酶催化的应用具有悠久的历史,最早可以追溯到古人的酿酒技术[2]。随着科学的进步,我们对酶催化的过程有了更深入的理解,同时在“碳中和”的大背景下,酶催化也因其高效环保、条件温和以及高立体选择性等优点被广泛应用于医药、化工等各领域[3-5]。此外,基于生物底盘的异源生物合成也非常依赖于由一系列酶催化反应组成的生物合成路线的优化与设计[6]。因此,酶被视为生物制造领域的核心“芯片”,而酶反应的机制解析与优化设计是“芯片”升级换代的重要驱动力。
在酶反应机制解析方面,虽然当前通过实验和计算(如多尺度模拟方法[7]等)结合来解析酶的三维结构、功能及其催化反应机制越来越流行[8-10],但因为直接验证反应机理的实验手段有限,而QM/MM等多尺度模拟的计算代价仍然较为昂贵,当前人们所探索的酶促过程只是酶反应空间中的冰山一角。而随着测序技术的发展,有大量酶序列的功能有待阐明[11],现有天然产物数据库也是日益丰富,但其中大量结构的生物合成反应路线仍有待解析[12],这些都严重制约了新酶的发现与天然产物的生物制造。在酶反应优化设计方面,尽管AlphaFold2[13]等蛋白结构预测工具为从一维序列到三维蛋白结构的理论预测提供了利器,但是基于序列的酶功能预测以及以功能为导向的蛋白序列设计相关算法进展则相对更滞后[14-15]。此外,在工业酶领域,如何拓宽酶的底物谱、改善酶反应选择性、提升酶催化效率或稳定性是重要的研究方向[16],但目前这些研究在很大程度上仍然依赖于研究人员的知识和经验。而当前广泛采用的多轮次“设计-构建-测试-学习(DBTL)”循环策略,往往要消耗大量的时间和资源。
随着大数据时代的到来,利用计算机从已知的各类数据中挖掘背后隐藏的序列与酶反应相关性成为可能。例如,合成路线与酶功能的计算预测[17-18]可助力于生物合成途径的设计与优化,而基于代谢组和基因组数据的代谢网络模型[19]以及全细胞模型[20]则可以对物种或细胞的代谢生长过程进行模拟,进而对上述设计路线进行计算测试。总之,近年来这些数据驱动的模型正在逐渐深入参与到传统DBTL的各个环节中,从而加速DBTL循环而缩短时间周期,抑或代替实验环节来缩减实验成本[21-22]。
基于上述现状,本文首先整理了常用的酶反应数据库,然后以反应底物、产物和酶为三个抓手对近年来酶反应预测和设计的计算工具进行了梳理,最后对数据驱动的酶反应预测与设计研究进行了展望。
1 酶反应数据库
在数字信息的时代,数据就是生产力,因此生物信息研究领域出现了许多高质量的数据库,不仅为传统的实验人员提供了信息服务,更是在数据驱动的计算工具开发中发挥了关键作用。表1汇总了常用的几个酶反应相关的数据库,这些数据库都有相应的Web服务器,可以直接在线访问和检索,并且除了Reaxys[32]外,其他数据库都可以免费下载使用。

表1 酶反应数据库Table 1 Databases of enzymatic reactions
在天然产物代谢领域常用的数据库为KEGG[23]和MetaCyc[24],两个数据库中均搜集了大量的酶反应,并且以生物合成途径对反应进行了不同层级结构的注释,如MetaCyc中针对次级代谢产物生物合成中划分有萜类生物合成途径、聚酮生物合成途径等,而萜类合成途径中又有单萜生物合成途径、萜类生物碱合成途径等等。Rhea[25]是由瑞士生物信息学研究所建立并维护的专门针对酶反应的数据库,其共同参与维护的还有蛋白序列数据库Uniprot[33],因此Rhea中的反应具有全面的酶信息注释,且与Uniprot高度关联。BRENDA[26]和SABIO-RK[27]则是致力于搜集酶反应动力学信息的数据库,包括米氏常数(Km)、催化常数(kcat)以及酶反应条件如温度、酸碱度(pH)等,而且BRENDA还提供了酶的详细分类(EC number等)和命名信息。Reactome[28]、PathBank[29]、HMDB[30]是具有不同侧重点的生物通路数据库,它们搜集了包括各种代谢反应、信号转导在内的各种信号通路数据。基于以上众多数据库各有侧重,但同时又有大量重复数据的情况,Pagni等[31]对KEGG、MetaCyc、HMDB等12个数据库的反应和酶进行汇总去重,构建了MetaNetX数据库,可用于基因组尺度的代谢网络模型的构建和分析。除了上述开源数据库以外,也有一些商业数据库可提供信息的检索和下载服务,如Elsevier旗下的Reaxys[32]数据库,包含了从各种专利和文献中提取的有机反应和酶反应数据。
2 酶反应预测与设计
反应底物、产物和酶是认知酶反应的三个核心要素,因此大部分酶反应的计算预测和设计方法都围绕这三点展开,且计算模型通常是通过其中之一(或之二)对剩余要素进行预测(图1):围绕底物、产物的正向或逆向预测探索反应和代谢物空间,同时还能用于合成路线的预测;根据给定反应预测所需的酶,或者反过来对未知反应功能的酶进行酶功能分类或反应活性强度预测;根据反应和酶的信息对催化反应重要性质(如反应动力学参数)进行预测等。因此,接下来论文将以酶、底物和产物为酶反应的三个抓手,从酶反应的数据表征、酶反应路线的正逆向预测、未知酶功能的预测与设计、已知功能的酶反应性质预测等方面来分别介绍。
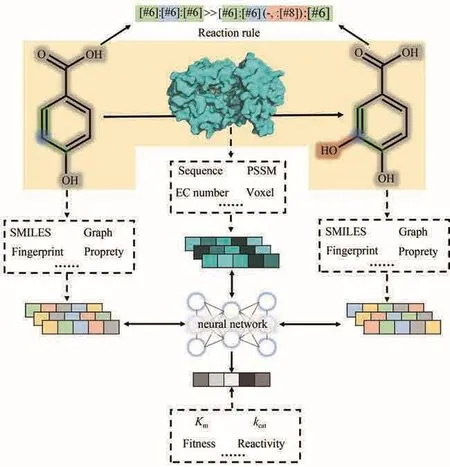
图1 酶反应的三个核心要素(底物、酶和产物)及其信息表征方式Fig.1 Key elements (substrate, enzyme and product) of enzymatic reactions and their information representations
2.1 数据的表征
在构建计算模型之前,我们需要对数据(即小分子和蛋白质的结构与性质)进行表征,使其转化成计算机能够理解的语言。无论是小分子还是蛋白质,都有不同维度的表征方式,如对于小分子来说,有基于二维结构的SMILES表达式、分子图(graph)和分子指纹等,还有基于三维结构的像素表征等[34-35],此外也能通过分子的一维理化性质如分子量、疏水性、电荷等进行表征[36]。对于蛋白结构来说,最常用的是一维的氨基酸序列表征,以氨基酸序列为基础的多序列比对(MSA)结果同样也可以作为表征。近年来多种蛋白质结构预测模型都表明MSA中序列共进化信息对于模型的预测精度有显著提升[37]。除此以外还能用二维的位置权重矩阵(PSSM)、接触图(contact map)、三维的像素点等对蛋白进行表征[17]。而对于化学反应,在深度学习模型发展起来之前,研究人员主要通过经验和知识对反应规则进行总结,并主要通过SMIRKS表达式(SMILES的一种拓展)来表示,其中包含了特定的反应位点信息和化学键的形成和断裂模式,一些常用的化学信息学工具如RDKit[38]等可以直接读取SMIRKS并将其应用于给定底物,从而判断其是否符合该反应规则并生成特定的产物。对于酶来说,其功能可直接由其催化的反应来表征,但除此以外,酶的分类学标签和基因本体论(gene ontology,GO)[39]注释也常用于描述酶的功能。酶的分类学标签通常指酶学委员会(Enzyme Commission)为酶所制作的一套编号分类法,该分类以化学反应的类型为基础。每个酶的EC number都由字母“EC”和四个数字组成,其中四个数字用点分隔,第一个数字使用数字1到7分别代表目前划分的七大类酶(氧化还原酶、转移酶、水解酶、裂解酶、异构酶、连接酶和转位酶)。后面三位数字将酶的分类逐级细分,由于不同大类下的子类数目不一,因此后三位数字的取值范围并不固定。而GO注释则是现代生物学从三个方面(分子功能、细胞组分、生物过程)对基因(及其表达的蛋白或RNA)所进行的描述。和EC number类似,每个方面之中又有各种细分的描述,一般称为GO term,如“GO:0005737”是细胞组分中的细胞质,表示某基因的产物是细胞质的组成成分。在机器学习模型中,数字表征(如分子量、电荷等)可以直接作为输入,而分子图、接触图等可转换为邻接矩阵进行输入,对于文本表征(如SMILES、氨基酸序列等)则有多种输入方式,如独热编码(one-hot编码)、词嵌入(word embedding)等。上述表征方式所提取出的特征各有侧重,因此在实际应用中通常需要根据任务的性质采用不同的表征方式进行模型训练。
2.2 基于反应物(以及酶)的产物预测
目前在自然界中仍然存在着大量未知的代谢过程,被称为“代谢暗物质”,阐明这些未知的代谢物和代谢反应能为新药发现和构建细胞工厂提供丰富的资源[40]。因此有许多工作聚焦于拓展现有分子的反应空间,即基于已知分子预测其潜在的各种代谢产物[图2(a)]。以Hatzimanikatis课题组[40]的工作为例,他们将前期总结的约500条反应规则[41]应用于150万个生物来源小分子及活性小分子,构建了ATLASx数据库。该数据库中一共包含了520万条和现有的8000万小分子有关的反应,且其中有148万小分子此前并没有包含在任意反应中,即为“孤儿”分子。ATLASx数据库极大地丰富了代谢反应空间,同时也为许多未知合成途径的化学分子指明了潜在的生物合成途径。作者利用该方法对上市药物诺斯卡品(noscapine)的生物合成途径进行了拓展,发现了另一天然来源的上市药物分子延胡索乙素(tetrahydropalmatine)的潜在的生物合成途径并在酵母细胞中构建该途径并验证了其正确性[42]。Hu课题组[43]从文献中搜集了28万条反应数据并提取出其反应中心及其相邻原子的变化作为反应规则,并利用反应指纹(即底物的分子指纹减去产物的分子指纹)对上述反应规则进行去重,基于此反应数据库开发了BCSExplorer工具用于探索给定分子的合成或代谢空间。另外还有基于传统分子相似性的方法从数据库中查找已有的反应对目标分子反应空间进行探索(表2)。

图2 正向和逆向反应预测[正向和逆向反应预测都是从一个分子(绿色)出发预测其潜在底物或产物(黄色),箭头表示两者之间能够通过反应进行转化,在(a)中箭头从反应物指向产物,(b)中则相反。经过多次迭代能够获得一个反应网络,网络中既能采样到已知的分子(实心)又能获得全新的结构(空心)。但不同的是正向反应预测每一次迭代方向都是随机的,而逆合成预测通常有一个终点条件(蓝色,如特定的原料分子),且会采取算法使得迭代过程朝着终点的方向进行]Fig.2 Prediction of forward and backward enzymatic reactions[Prediction starts with an enzyme molecule (green node) to deduce its substrate or product (yellow nodes), the lines represent transformation reactions between two molecules, with arrow from substrate (enzyme) to product (a) and the reverse (b).A reaction network is developed after the iterative prediction in which both known (solid nodes) and unknown (hollow nodes) molecules are included.The forward prediction is generally random while a target (blue node, such as a building block) is specified in the backward prediction, and the exploration will lead to the target with the help of iterative algorithms.]

表2 酶反应预测与设计工具汇总Table 2 Tools for the prediction and design of enzymatic reactions
除了上述传统的方法,Reymond课题组[45]利用SMILES和文本编码分别表征小分子和酶,将其用于深度学习模型Transformer[66]的输入,从而对产物进行预测。为了克服酶反应数据量不足的问题,作者采取了迁移学习的策略,先利用大量有机合成反应对模型进行预训练,再利用酶反应继续训练。研究结果表明有机反应的预训练确实对模型最终的预测能力有提升,并且和只使用反应物信息相比,酶信息的加入也有助于模型做出更加可信的预测。利用该模型不仅能对酶催化的产物做出预测,还能对酶的底物谱进行筛选,进一步阐明酶的催化功能。Kavraki及其合作者[46]则是利用深度学习构建了一个专门预测药物在人体内潜在代谢产物的预测模型,该模型同样是以底物SMILES作为输入,但不同的是该模型并没有包含酶的信息,因为对于药物代谢来说,所有可能的产物比特定酶催化得到的产物更加有指导意义,且作者测试发现在酶的信息数量有限的情况下,包含酶的信息对于模型的提升并不大。
2.3 基于产物的逆合成预测
和上述正向预测相比,逆合成预测[67-68]具有更强的目的性,它是对特定化合物的合成前体进行预测并将该过程循环迭代直到到达终止条件(如路线找到了一些常见的合成前体或容易获得的化学原料等)。由于逆合成预测的任务通常是找到目标分子和特定合成前体之间的合成路线,因此在每一步预测时并不会像正向预测那样任意拓展,而是需要进行评估和筛选以节约计算资源[图2(b)]。尽管如此,为了避免错过“正确”的合成前体,每一步逆合成预测依然会输出不止一个可能的结果,最终的路线组合数量会随着迭代步数增加呈指数级增长,因此在逆合成预测过程中仍然需要配合高效的搜索算法对路线分支进行“修剪”。Faulon团队[69]从MeteNetX反应数据库中自动提取出了超过上万条反应规则,并将其应用于生物逆合成路线的预测。由于许多分子可以同时应用多条反应规则,因此每一步逆合成预测都会产生大量的候选前体分子,为了从巨大的组合空间中高效搜索潜在的合成路线,作者首先采用了结构相似性和可用的蛋白序列数量对每一步结果进行打分,并结合蒙特卡洛树搜索[70]的策略优先选择更加可靠的前体分子进入后续的迭代预测[48]。为了使每一步的预测更加可靠,Turner及其合作者[49]则开发了RetroBioCat工具,通过人工总结常用的生物催化反应并编码反应规则,将其应用于生物催化级联合成路线的预测和设计,该工具很好地重现了文献报道的五十余条生物催化合成路线。
此外,深度学习模型凭借其无需构建反应规则就能捕捉反应信息的优势,也逐渐被应用于逆合成的预测[71]。Wu及其合作者[50]搜集了天然产物合成相关的3万余条反应并利用SMILES进行编码,用于天然产物生物逆合成模型BioNavi-NP的训练。作者还从有机反应数据中提取出了6万余条和天然产物结构类似的反应用于数据集的扩充并进行迁移学习,测试结果发现基于Transformer结构的模型表现要好于普通的神经网络和基于反应规则的模型。同时作者采用了基于与或树的Retro*搜索算法[72]用于多步反应路径的搜索,经过测试表明,该方法速度和精度远好于蒙特卡洛树搜索。该工具不仅可用于天然产物生物合成路线的预测,还能对已有的生物合成路线进行重构,有助于寻找更加高效的异源合成途径。Probst团队[51]也采取深度学习的方法,利用现有的酶催化反应分别将底物和产物作为模型输入构建了正向和逆向合成两个模型,在正向预测模型中酶的EC number也同时作为输入,而在逆合成预测中EC number则是作为输出,因此在逆合成模型中不仅能输出目标化合物的前体,还能对所需酶的类别进行预测。
上述模型能分别对代谢物和前体空间进行探索,但由于训练数据侧重的差异,不同模型有各自的应用范围,如药物代谢模型[46]仅能用于特定细胞色素P450酶的产物预测,RetroBioCat[49]和BioNavi-NP[50]则由于其训练数据的各自选择偏好与不足限制了其特定的一些适用范围。此外,和基于反应规则的方法相比,基于深度学习的方法虽然无需构建反应规则,但反应规则中的酶信息也同时被忽略了。尽管Probst团队[51]在模型中加入了EC number信息,但最多只能给出其前三位分类,对酶的预测仍然有限。
2.4 基于特定功能的酶搜索和设计
尽管正向和逆向反应模型在针对特定体系的预测中都有不错的精度,但在许多模型中酶的信息并没有被充分考虑,限制了其应用范围。在实际应用尤其是逆合成预测中获得潜在的合成途径之后,催化相应反应的酶对异源表达至关重要。尽管目前已有众多方法和平台可以用于酶蛋白元件的挖掘[73],但获取催化特定反应的酶仍然是一个艰巨的任务。目前解决此类任务的思路主要是相似的反应往往可以通过同一个酶来催化完成,因此很多工具都是从已有数据库中搜索和目标反应相似的反应[图3(a)],以催化该相似反应的酶作为候选序列进行后续的实验和改造。如ECBLAST[52]可以通过键的变化、反应中心以及结构的相似性从KEGG数据库中寻找相似反应,并给出相应酶的EC number。Selenzyme[53]是一个基于结构相似性的酶搜索工具,用户输入反应规则或一条具体的反应后,Selenzyme会在MetaNetX反应数据库中搜索相似的反应,并以催化该相似反应的酶作为结果输出。用户还可以对所需酶的物种做出限制,该工具将会根据候选酶所在的物种与用户指定物种之间的系统发育距离对结果重新排序。其他工具如BridgIT[54]、E-zyme2[55]则分别通过反应指纹和反应模式对KEGG数据库进行搜索,从而对目标反应进行酶的预测。
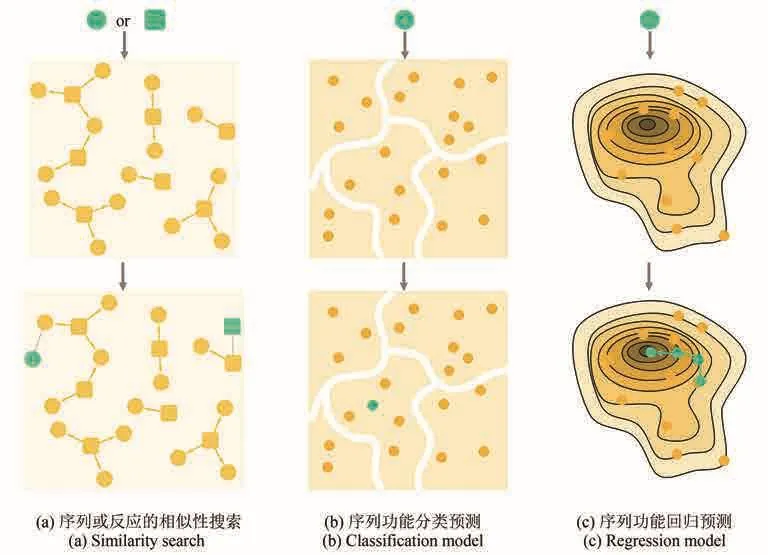
图3 不同类型的酶搜索和功能预测模型。[圆形代表酶,方形代表反应,黄色节点代表已知数据,绿色代表待预测对象。基于相似性的预测方法(a)是从已知的酶-反应数据对中(图中相连的两个节点)寻找与目标对象相似的样本,从而对其反应(或酶)进行预测。功能分类模型(b)是将已知功能(通常是离散变量)的酶序列作为训练集,寻找其潜在分类规律(白色分界线),从而对目标序列进行预测。回归模型(c)则是对活性或稳定性强弱等连续变量进行建模预测,绘制适应度景观图从而对目标序列的功能进行预测,并用于酶设计]Fig.3 Models for searching and predicting enzymes[Circular and square nodes represent sequences and reactions, respectively, and yellow filling indicates known data while green filling mean objects to be predicted.Similarity search (a) is to find a similar object in known enzyme-reaction pairs (connected nodes) to predict reactions (or enzymes)for target object.Classification model (b) is trained by enzymes with known function (usually discrete), in which the classification rule (white boundary) is clarified, and then the model can be used to classify an enzyme with unknown function.Regression model (c) is adapted to draw fitness landscape to predict continues variables such as the activity or stability of enzymes, which can then be used for enzyme design.]
除上述基于反应相似性的方法以外,近年来基于机器学习的工具也展现出潜在的应用价值。当前由于非冗余酶反应数据的不足,单纯依靠已知反应来寻找所需的酶难度极大。最近Faulon团队[56]采用了同时给定反应和酶,进而判断该酶是否能催化该反应的思路。作者利用反应中心原子环境的变化和氨基酸序列对反应和酶分别进行表征,将两者共同输入高斯过程分类模型进行训练。由于现有数据库中只有某种酶能够催化某些反应的数据(即正样本),而缺少负样本,因此作者有选择地从不同酶-反应数据对中分别挑选酶和反应组成新的样本,并作为无标签样本进行半监督学习。作者利用该模型挑选出了大肠杆菌中和N-乙酰-L-亮氨酸合成相关的酶并经实验成功验证。
虽然酶是高效、精准的催化剂,但大部分天然的酶却很难直接应用于工业生产[15],传统的突变扫描实验尽管可以获得潜在的功能序列,但通常需要多轮次的实验筛选,不仅耗时长且成功率低。而数据驱动的酶计算改造和重设计可以绕过实验的突变筛选,对潜在的序列和活性空间进行高效搜索。如Ranganathan及其合作者[57]将分支酸变位酶(chorismate mutase,参与芳香族氨基酸生物合成的关键酶)的MSA信息用于玻尔兹曼机器(Boltzmann machine)学习的直接耦合分析(direct coupling analysis,DCA)[74],得到的模型可捕捉序列的保守位点和位点之间的相关性,因此可用于突变体的采样。近年来,蛋白质的从头设计(de novodesign)[75]在深度学习的辅助下也取得了一些进展,诸如Baker课题组[76]和Liu课题组[77]都分别基于深度学习框架提出了逆向折叠(即利用蛋白结构生成序列)算法,有望针对催化特定反应的结构或活性位点设计出全新的蛋白序列[78]。
2.5 酶功能预测
随着测序技术的进步,数据库中有大量的序列已知但功能未知的待定蛋白元件,因此亟需可靠准确的方法对这些蛋白序列进行功能预测。由于只有序列信息已知,因此传统的方法便是通过序列相似性从数据库中搜索已知功能的蛋白(如BLAST[79]),从而对目标蛋白的功能做出预测[图3(a)]。而机器学习方法由于能够从更多特征中学习隐藏规律,更有可能取得精确的预测结果。目前针对酶功能最常用的标签是EC number,有许多机器学习方法通过序列进行EC number的分类预测[图3(b)]。如Lee课题组[58]开发了深度学习模型DeepEC,利用卷积神经网络对氨基酸序列进行编码并预测其EC number。经测试DeepEC比其他5种预测工具具有更高的准确率和更快的运行速度。Araki团队[59]则通过PROFEAT工具[80]从酶的氨基酸序列中提取性质特征(如氨基酸组成等),从而对其进行编码,然后输入机器学习分类模型中预测其EC number。测试结果显示支持向量机(SVM)模型的表现要优于随机森林(RF)、k-邻近算法(kNN)和多层感知机(MLP)模型。作者利用该模型对罂粟中苄基异喹啉生物碱合成路线中缺失的酶进行预测,成功解析了该物种中酪氨酸到下游生物碱的分支合成途径[81]。除了EC number以外,GO条目也常被用于蛋白功能预测模型中。Jones课题组[60]构建了一个多任务深度神经网络(MTDNN)用于预测给定序列的GO注释,其中多任务表示除了整个模型共有的隐藏层以外,对于每一个GO条目都有独立隐藏层网络来负责输出最后的预测结果。MTDNN采用了多种功能和结构描述符来编码蛋白序列如二级结构、跨膜组分等。MTDNN的预测精度不仅高于传统的BLAST方法,也比单纯的多标签分类神经网络要好。
和上述普适的功能分类预测不同的是,在蛋白质工程中,往往需要针对特定功能的酶进行活性强度的预测,即回归模型[图3(c)],并利用该模型对突变体进行预测从而在多轮的突变实验中以筛选出最优序列。ECNet[61]是一个基于序列共进化信息进行活性预测的神经网络模型,蛋白序列表征由基于大数据库(如Uniprot[33]或Pfam[82])预训练得到的全局特征和基于目标序列MSA得到的局部特征组合而成。将已有的深度突变扫描实验数据对模型进行训练,可以对未知的突变体活性进行准确预测,作者以β-内酰胺酶为例成功从模拟突变中筛选出活性强于野生型的新颖突变体。Gitter团队[62]则利用接触图作为蛋白表征,同时采用图卷积神经网络对蛋白活性进行预测。作者将随机重启爬山算法和该模型结合从链球菌蛋白G的GB1结构域(能与免疫球蛋白G结合,可用于抗体纯化)序列空间搜索得到了5个高亲和力的突变体,经实验测试,其中一个突变体确实表现出比野生型更强的结合亲和力。该类模型能够代替传统实验中的构建与测试环节,减少的DBTL循环迭代次数,从而针对特定的酶快速绘制出清晰的序列-功能图谱,将其应用于定向进化等蛋白质工程任务中可以极大地节约筛选时间和实验成本[83-84]。
2.6 酶反应性质预测
酶反应动力学研究对于理解酶反应机制,以及设计合理的反应条件具有重要作用[85-86]。其中Km和kcat是衡量酶催化效率最重要的两个参数,而实验是获得这些参数的主要手段。近年来,有不少工作利用深度学习模型来对酶反应动力学参数进行预测。如Lercher及其合作者[63]利用分子指纹、脂水分配系数以及分子量表征反应底物,用一个经过大量已知蛋白序列预训练过的特征提取模型UniRep[87]对酶进行表征,并使用梯度提升构建回归模型用于预测反应的Km值。在独立的测试集中该模型也表现出良好的性能。此外,也有模型专门预测同一物种中同一个底物与不同酶作用时的Km值[88],以及纤维二糖在不同β-葡萄糖苷酶催化下的Km值[89]。Palsson及其合作者[64]利用代谢网络、蛋白结构、物质浓度等多种信息输入机器学习模型(包括随机森林、深度神经网络等)进行大肠杆菌代谢网络中酶反应kcat的预测。尽管通过该模型对kcat进行预测能够提高代谢网络模型的精度,从而对细胞生长状态进行更准确的预测,但其输入数据处理太复杂,很难应用于其他物种。因此Nielsen课题组[65]从BRENDA和SABIO-RK数据库中搜集了所有带有kcat注释的酶反应,并用底物的分子图进行编码,以及用氨基酸序列对酶进行编码,两者分别用于图神经网络和循环神经网络的输入,并在拼接之后用于kcat的预测。该模型除了在测试集中表现优越之外,对于酶序列具有细微变化的突变体的催化能力预测也有不错的准确率,且神经网络中注意力机制的加入能够让模型检测到和酶催化效率相关的关键残基。为了数据共享并服务于更多研究者,作者基于该预测模型的结果构建了一个在线的酶反应参数数据库,在目前的版本中可对计算机预测的反应kcat值进行查询[90]。无论是Km还是kcat或是其他酶反应动力学参数,通过实验上的测量都有一定的难度,且受实验条件影响较大,因此计算模型的出现提供了巨大的便利。并且,这些参数对于目前各种类型基因组尺度的代谢网络模型[19]以及全细胞模型[91-92]至关重要,在未来,有望利用这些模型对物种或细胞整个生命周期进行模拟,不仅能让我们更好地理解生物体内的生长状态和生理过程,更能帮助我们设计和优化人工底盘细胞用于生物合成。
3 展 望
尽管随着各种实验技术的进步和计算模拟技术的引入,大自然酶催化这层神秘面纱正一点一点被揭开,但是在此面纱之下还有巨大的未知空间有待挖掘。以酶为核心的生物制造技术是极具潜力的发展方向。挖掘更多的酶催化元件、优化酶催化的功能与效率,提升工业酶的性能,是生物经济产业发展的重大需求。尽管蛋白结构的理论预测获得了重大突破,但酶催化机制的高效解析、酶功能的理性设计等仍然是领域内的难点。从蛋白的静态结构到酶反应的动态调控机理,从海量而冗余的酶反应数据到大数据驱动的酶分子设计,这中间还有不少鸿沟需要新理论、新技术的突破。本文旨在从大数据驱动的酶反应预测与设计的视角,汇总当前常用的酶反应数据库,并对近年来基于大数据和机器学习开发酶反应预测与设计工具进行了总结。上述一些成功的案例已经预示着未来,计算模型与算法的发展将是走向生物智造不可或缺的核心推力之一。然而我们也必须清醒意识到,现有计算工具在精度上还有很大的提升空间,要最大化地发挥计算的赋能作用还需要不断地探索(图4)。
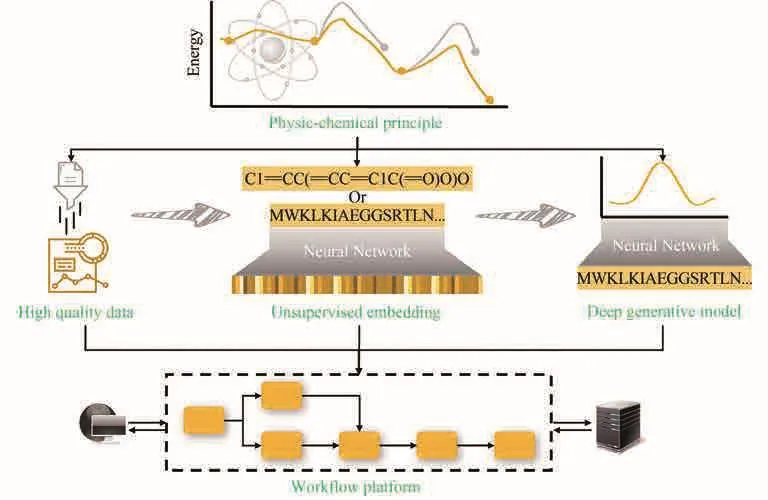
图4 数据驱动的酶反应设计与预测在未来的展望Fig.4 Perspective of data-driven prediction and design of enzymatic reactions
首先,尽管当前已经有了诸多类型的数据库,但这些数据来源于世界各地的实验室,其实验条件、实验试剂等都会对实验数据造成影响,例如大多数酶的反应动力学参数对于实验温度、pH等都是敏感的[93]。幸运的是现有数据库中已经开始对这些实验条件进行了记录,而如何将这些条件纳入计算模型的构建中将是我们下一步要思考的问题[94]。此外,不管数据库的标注和更新是由人工还是计算机完成,都不可避免会有错误记录的产生,有文献就指出BRENDA数据库中有相当一部分酶的EC number注释是错误的[95]。因此尽可能避免错误标注,提高数据的质量是提升计算模型精度很重要的手段。此外,对于很多机器学习模型而言,训练集中负样本对于模型优化是极为重要的,但当前数据库收录的都是文献中报道的“阳性”结果(即能反应或有活性),而经实验验证的阴性结果同样包含重要信息,因此方便用户上传实验结果(无论是阴性还是阳性)的数据共享平台的建设也值得我们关注,当然这也需要领域内实验人员共同的努力。另外,智能化机器人也能在很大程度上解放实验人员,同时可以完成实验、记录、保存等一系列工作,加快正、负数据的积累从而加速实验与计算交互反馈的进程。
其次,无论是针对小分子还是蛋白的机器学习特征提取,我们都无法用数字解释其包含的所有信息,尽管我们也并不一定需要这样做,但是如何尽可能提取出我们所需要的输入信息并进行编码,也有待于进一步研究。近几年来自然语言处理模型的进步使我们能够利用非监督学习的方式从大量语料库(比如分子库或蛋白序列库)中训练出用于输出小分子和蛋白的嵌入表征的预训练模型[87,96-98]。经分析这些模型能够很好地捕捉不同样本的潜在特征并用于输入,下游针对各种任务的机器学习模型性能有望从中得到提升[99]。
最后,在计算模型与算法层面,同样也是得益于自然语言处理模型的发展,越来越多生成式的端到端模型应用于反应预测和设计领域[71,100]。和普通的回归或分类模型以标签为输出不同的是,端到端模型可以直接通过训练过的概率分布生成序列(在本文中即小分子或蛋白)。如前文提到的酶催化反应的正向和逆向预测的模型中,有不少就已经应用了该种端到端模型[45,50]。而随着各种蛋白序列生成模型的提出[101],将其应用于酶的预测将会大大扩大搜索空间,因为目前基于已知反应的酶预测还局限于从蛋白序列库中寻找潜在的酶或其突变体,生成模型则能在特定限制条件下产生具有潜在功能的新颖序列,此外还能生成更多的非天然序列用于蛋白优化与设计[102-105]。
以上是围绕数据驱动的策略在将来需要重点关注的三个方面,而对于酶反应来说,其背后的物理化学原理对于酶反应设计是至关重要的,对于酶反应物理化学规律的认知信息在上述数据驱动策略中往往被忽略或者未考虑周全。在过去的十几年中,基于物理模型的方法在酶反应预测和设计方面已经取得了许多成果。如基于量子力学和分子力学组合方法(QM/MM)的动力学模拟不仅用于探索分子的反应性,扩展反应空间[106-107],还用于解析酶反应的机理,并为酶改造和重设计提供了极为重要的酶反应热动力学属性和酶催化调控的理论依据[8,108]。以Rosetta Design[109-110]为代表的基于物理模型的计算方法则为酶设计提供了新范式,并成功应用于许多案例[111-113]。而数据驱动的方法尤其是深度学习模型近几年虽快速发展,但作为一种被称为“黑箱”模型的工具,目前仍无法参与到如微观机制解析这种复杂且动态的任务中。我们既需要依靠物理模型解决最底层的问题并积累更多的数据,用于数据模型的构建和训练,反过来数据模型因其高效预测能力使其能够参与到物理模型的框架中,实现优势互补。因此,物理模型与数据模型的结合将是酶反应预测和设计的新趋势,如结合动力学模拟和深度学习的反应空间探索[114]、借助深度学习势能的分子动力学模拟方法[115]、基于神经网络能量函数的氨基酸序列设计[77]等等。
最后,为上述模型与算法搭建高度集成的工程化平台也是非常有意义的。目前只有少数计算模型和工具发布了在线服务器版本,其他大部分都是发布于各托管平台的源代码,并且目前很多深度学习模型体量庞大,需要一定的硬件支持才能运行,使用这些工具对于普通的实验人员来说有一定的壁垒。同时本文提到的反应预测、酶预测、酶反应性质预测在实际的实验中通常是链条式的流程,因此将这些工具整合在一个便捷友好的平台中将会给实验人员带来极大的便利。Hu课题组建立的RxnFinder(http://www.rxnfinder.org/)商业化网站平台[116]开发并整合了反应探索、前体预测、逆合成分析、菌株设计等一系列计算工具,能为实验研究人员提供便利。Hatzimanikatis课题组和曼彻斯特大学精细化学品合成生物学研究中心分别搭建的LCSB(https://lcsb-databases.epfl.ch/Home)和SYNBIOCHEM(https://synbiochem.co.uk/)数据平台也包含了各自课题组开发的逆合成预测、酶选择等工具供学术界免费试用。但这些平台只是将工具汇总在一起,用户需要单独访问或下载使用某一模块,并且针对相似任务的不同算法部署在不同的平台,不利于用户进行直接比较研究。未来在以酶催化为基础的生物制造工业化应用中,我们可能更需要全链条式的设计平台,即所有工具以工作流的形式集成在平台中,用户可一键访问并自由组合使用。
